

Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits
- PMID: 15877281
- PMCID: PMC1196445
- DOI: 10.1086/430841
Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits
Abstract
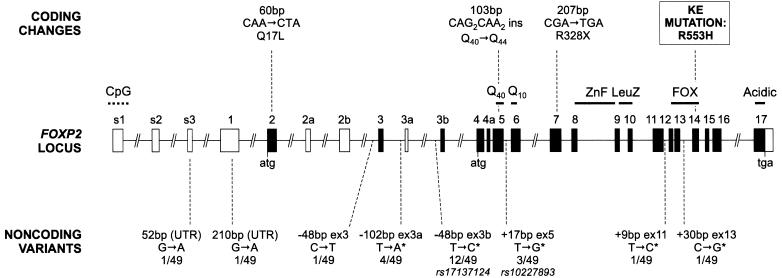
FOXP2, the first gene to have been implicated in a developmental communication disorder, offers a unique entry point into neuromolecular mechanisms influencing human speech and language acquisition. In multiple members of the well-studied KE family, a heterozygous missense mutation in FOXP2 causes problems in sequencing muscle movements required for articulating speech (developmental verbal dyspraxia), accompanied by wider deficits in linguistic and grammatical processing. Chromosomal rearrangements involving this locus have also been identified. Analyses of FOXP2 coding sequence in typical forms of specific language impairment (SLI), autism, and dyslexia have not uncovered any etiological variants. However, no previous study has performed mutation screening of children with a primary diagnosis of verbal dyspraxia, the most overt feature of the disorder in affected members of the KE family. Here, we report investigations of the entire coding region of FOXP2, including alternatively spliced exons, in 49 probands affected with verbal dyspraxia. We detected variants that alter FOXP2 protein sequence in three probands. One such variant is a heterozygous nonsense mutation that yields a dramatically truncated protein product and cosegregates with speech and language difficulties in the proband, his affected sibling, and their mother. Our discovery of the first nonsense mutation in FOXP2 now opens the door for detailed investigations of neurodevelopment in people carrying different etiological variants of the gene. This endeavor will be crucial for gaining insight into the role of FOXP2 in human cognition.
Figures

References
Electronic-Database Information
-
- dbSNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
-
- American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders (DSM-IV). American Psychiatric Association, Washington, DC
-
- Bruce HA, Margolis RL (2002) FOXP2: novel exons, splice variants, and CAG repeat length stability. Hum Genet 111:136–144 - PubMed
-
- Enard W, Przeworski M, Fisher SE, Lai CSL, Wiebe V, Kitano T, Monaco AP, Pääbo S (2002) Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418:869–872 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
