

Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction
- PMID: 15889149
- PMCID: PMC1142598
- DOI: 10.1038/sj.emboj.7600667
Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction
Abstract
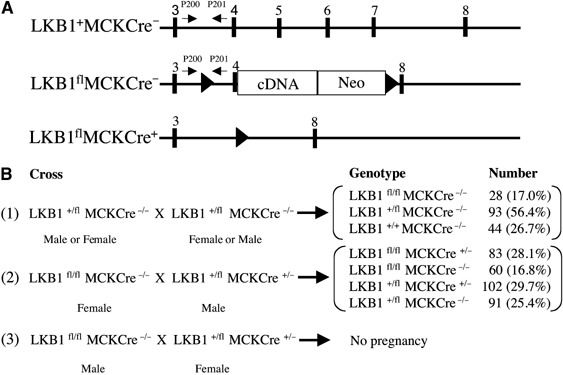
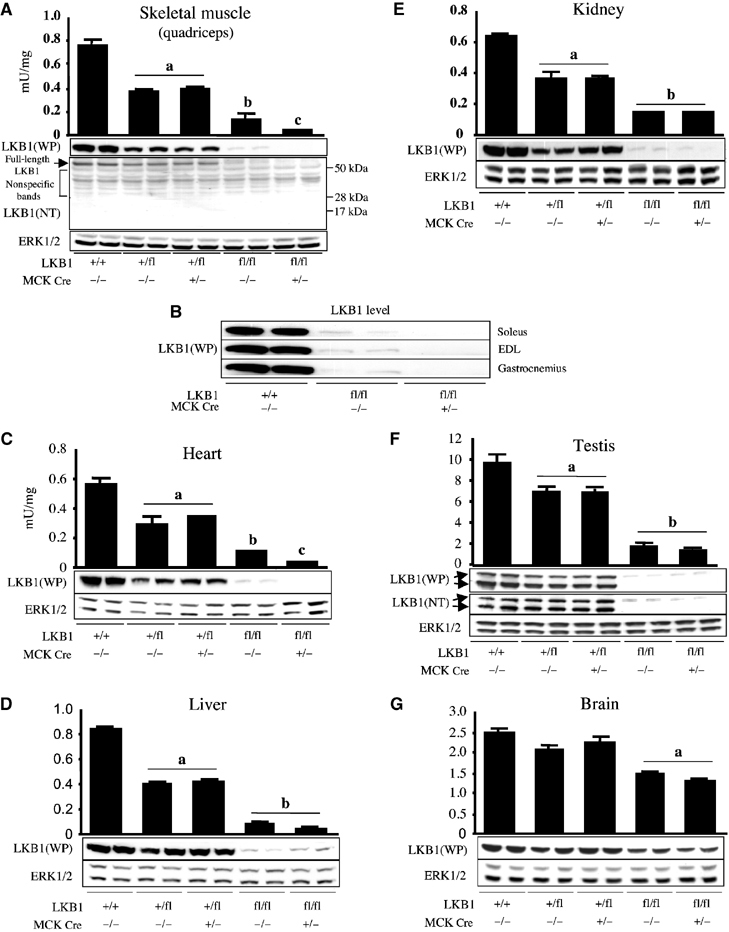
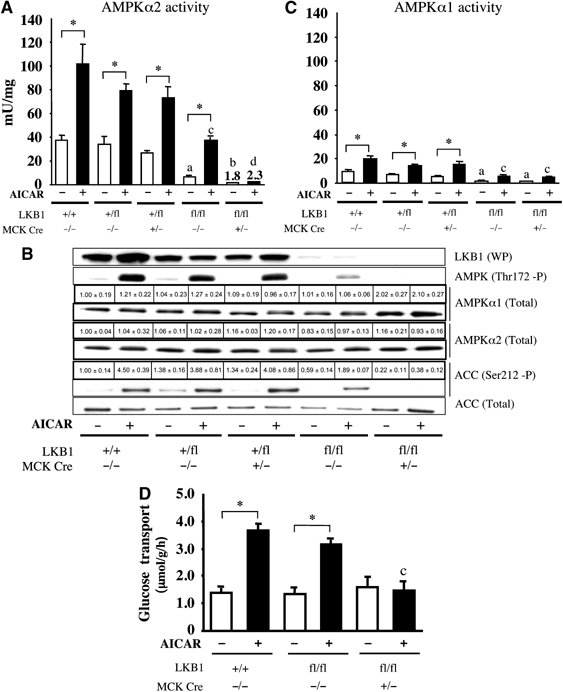
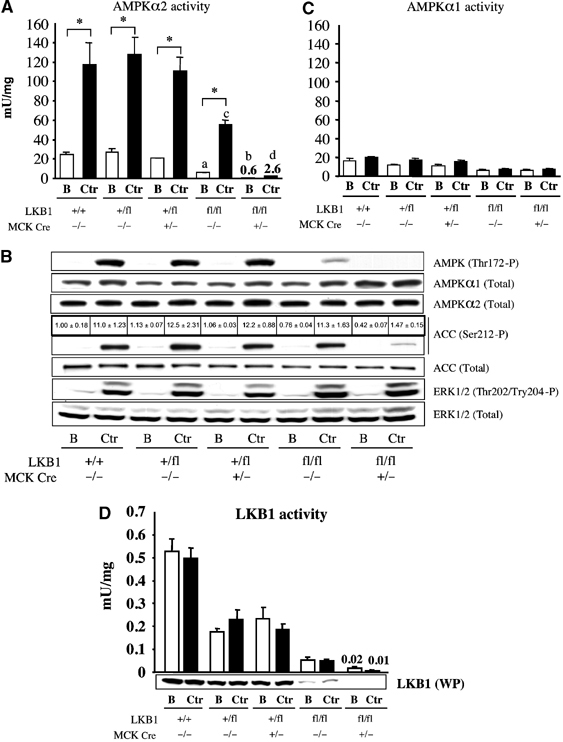
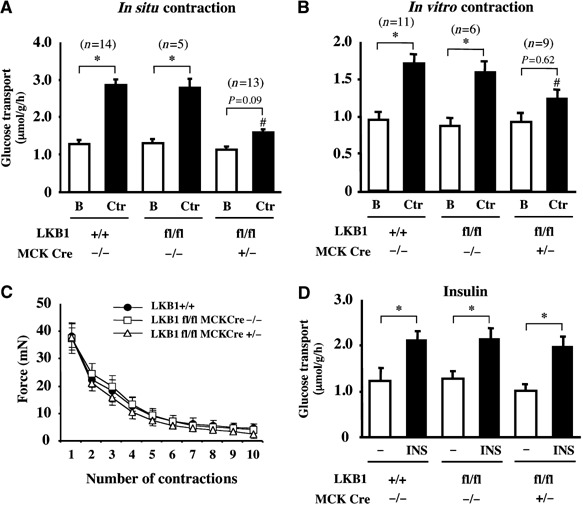
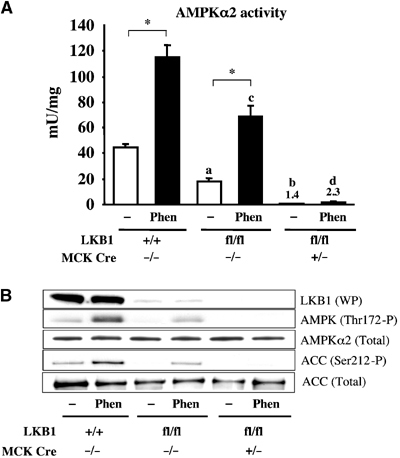
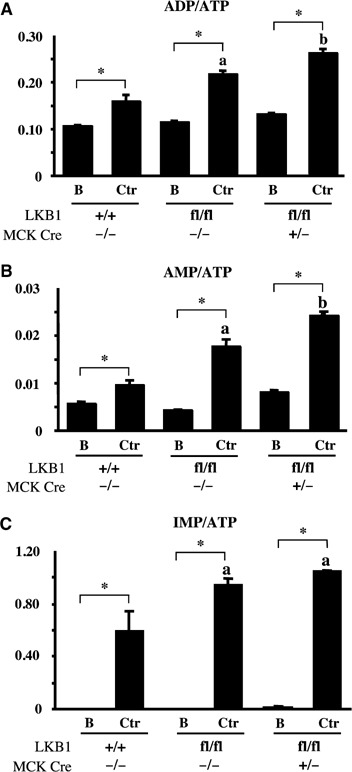
Recent studies indicate that the LKB1 tumour suppressor protein kinase is the major "upstream" activator of the energy sensor AMP-activated protein kinase (AMPK). We have used mice in which LKB1 is expressed at only approximately 10% of the normal levels in muscle and most other tissues, or that lack LKB1 entirely in skeletal muscle. Muscle expressing only 10% of the normal level of LKB1 had significantly reduced phosphorylation and activation of AMPKalpha2. In LKB1-lacking muscle, the basal activity of the AMPKalpha2 isoform was greatly reduced and was not increased by the AMP-mimetic agent, 5-aminoimidazole-4-carboxamide riboside (AICAR), by the antidiabetic drug phenformin, or by muscle contraction. Moreover, phosphorylation of acetyl CoA carboxylase-2, a downstream target of AMPK, was profoundly reduced. Glucose uptake stimulated by AICAR or muscle contraction, but not by insulin, was inhibited in the absence of LKB1. Contraction increased the AMP:ATP ratio to a greater extent in LKB1-deficient muscles than in LKB1-expressing muscles. These studies establish the importance of LKB1 in regulating AMPK activity and cellular energy levels in response to contraction and phenformin.
Figures
References
-
- Altarejos JY, Taniguchi M, Clanachan AS, Lopaschuk GD (2005) Myocardial ischemia differentially regulates LKB1 and an alternate 5′AMP-activated protein kinase kinase. J Biol Chem 280: 183–190 - PubMed
-
- Aschenbach WG, Sakamoto K, Goodyear LJ (2004) 5′ adenosine monophosphate-activated protein kinase, metabolism and exercise. Sports Med 34: 91–103 - PubMed
-
- Baron SJ, Li J, Russell RR III, Neumann D, Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA, Young LH (2005) Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res 96: 337–345 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
