

VEGF-integrin interplay controls tumor growth and vascularization
- PMID: 15897451
- PMCID: PMC1129024
- DOI: 10.1073/pnas.0502935102
VEGF-integrin interplay controls tumor growth and vascularization
Abstract
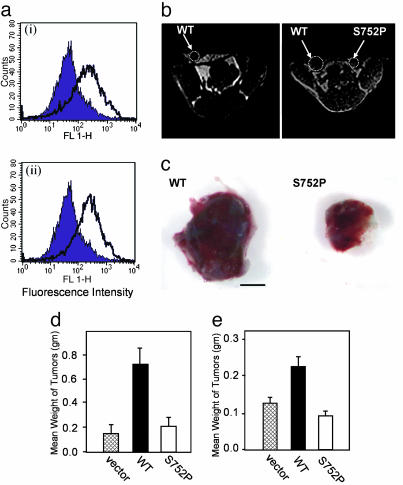
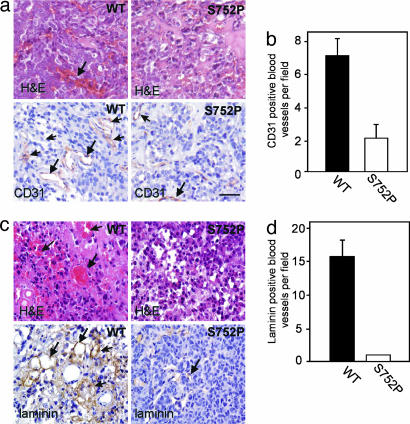
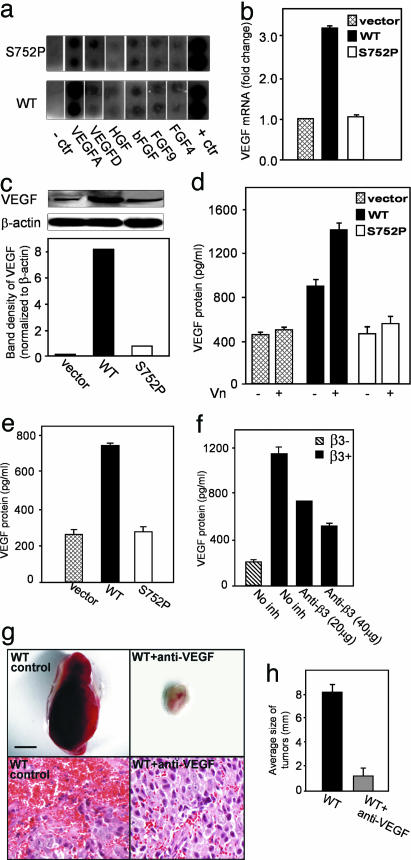
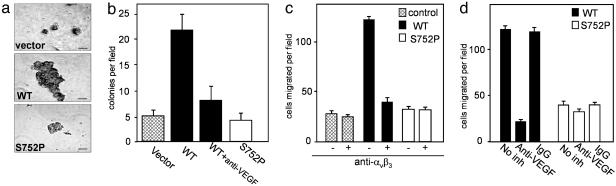
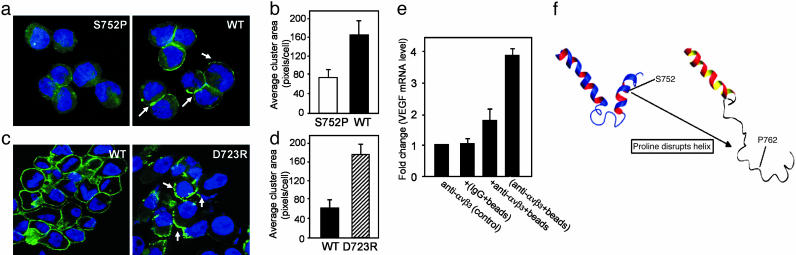
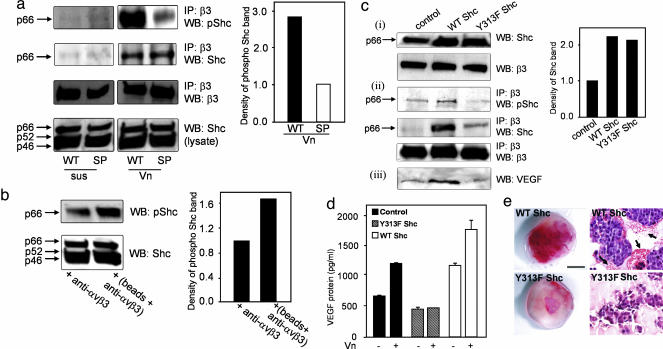
Cross-talk between the major angiogenic growth factor, VEGF, and integrin cell adhesion receptors has emerged recently as a critical factor in the regulation of angiogenesis and tumor development. However, the molecular mechanisms and consequences of this intercommunication remain unclear. Here, we define a mechanism whereby integrin alpha v beta3, through activation, clustering, and signaling by means of p66 Shc (Src homology 2 domain containing), regulates the production of VEGF in tumor cells expressing this integrin. Tumors with "activatable" but not "inactive" beta3 integrin secrete high levels of VEGF, which in turn promotes extensive neovascularization and augments tumor growth in vivo. This stimulation of VEGF expression depends upon the ability of alpha v beta3 integrin to cluster and promote phosphorylation of p66 Shc. These observations identify a link between beta3 integrins and VEGF in tumor growth and angiogenesis and, therefore, may influence anti-integrin as well as anti-VEGF therapeutic strategies.
Figures
References
-
- Neufeld, G., Cohen, T., Gengrinovitch, S. & Poltorak, Z. (1999) FASEB J. 13, 9–22. - PubMed
-
- Folkman, J. (2002) Semin. Oncol. 29, 15–18. - PubMed
-
- Ferrara, N. (2004) Endocr. Rev. 25, 581–611. - PubMed
-
- Varner, J. A. & Cheresh, D. A. (1996) in Important Advances in Oncology: 1996, eds. Devita, V. T., Hellman, S. & Rosenberg, S. A. (Lippincott Williams & Wilkins, Baltimore), pp. 69–87.
-
- Hong, Y. K., Lange-Asschenfeldt, B., Velasco, P., Hirakawa, S., Kunstfeld, R., Brown, L. F., Bohlen, P., Senger, D. R. & Detmar, M. (2004) FASEB J. 18, 1111–1113. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
