

Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins
- PMID: 15901672
- PMCID: PMC1142553
- DOI: 10.1101/gad.1304105
Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins
Abstract
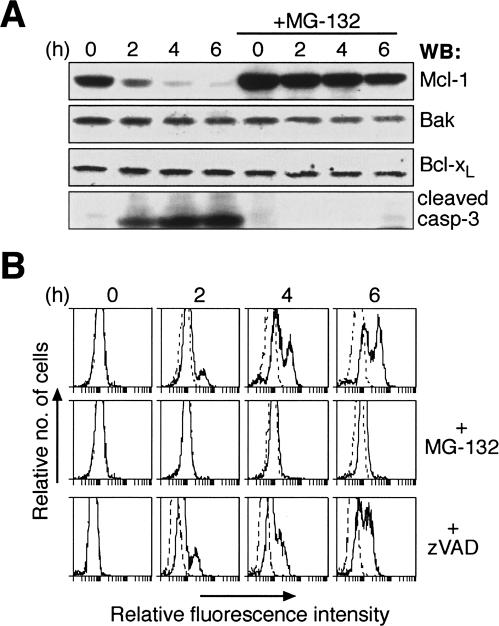
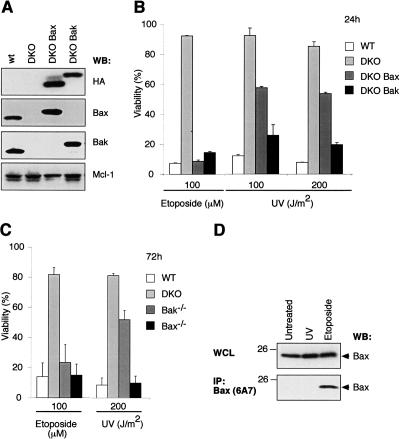
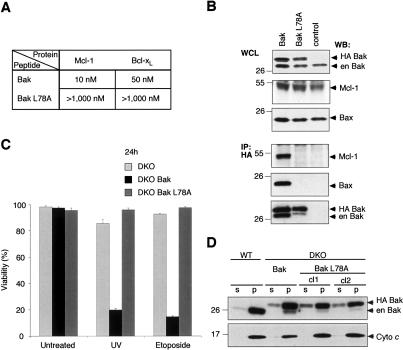
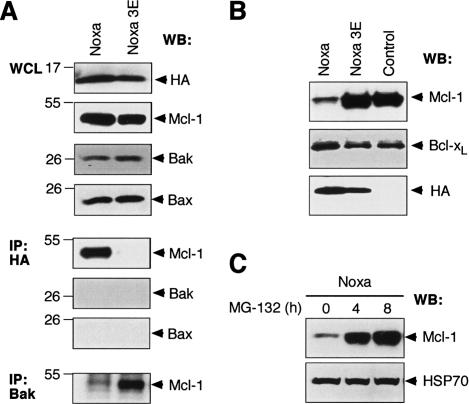
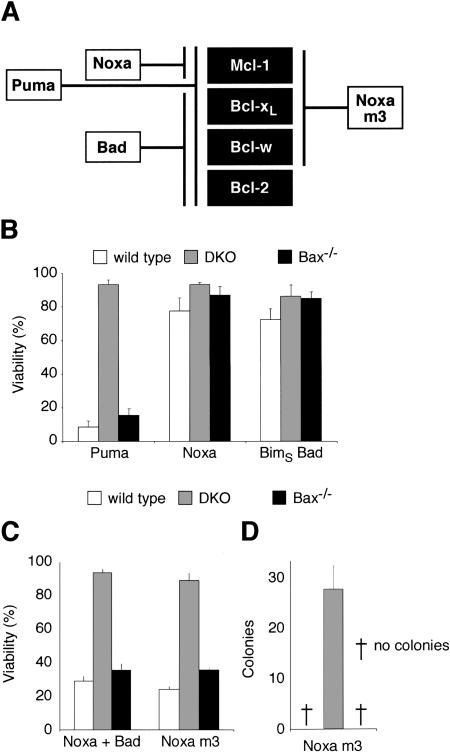
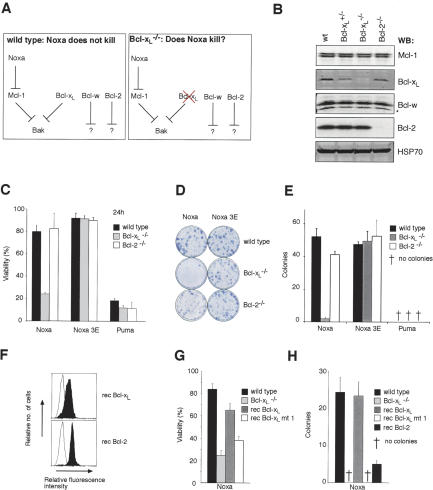
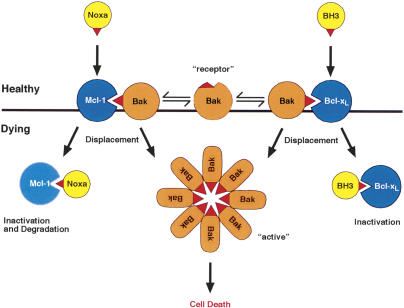
Commitment of cells to apoptosis is governed largely by the interaction between members of the Bcl-2 protein family. Its three subfamilies have distinct roles: The BH3-only proteins trigger apoptosis by binding via their BH3 domain to prosurvival relatives, while the proapoptotic Bax and Bak have an essential downstream role involving permeabilization of organellar membranes and induction of caspase activation. We have investigated the regulation of Bak and find that, in healthy cells, Bak associates with Mcl-1 and Bcl-x(L) but surprisingly not Bcl-2, Bcl-w, or A1. These interactions require the Bak BH3 domain, which is also necessary for Bak dimerization and killing activity. When cytotoxic signals activate BH3-only proteins that can engage both Mcl-1 and Bcl-x(L) (such as Noxa plus Bad), Bak is displaced and induces cell death. Accordingly, the BH3-only protein Noxa could bind to Mcl-1, displace Bak, and promote Mcl-1 degradation, but Bak-mediated cell death also required neutralization of Bcl-x(L) by other BH3-only proteins. The results indicate that Bak is held in check solely by Mcl-1 and Bcl-x(L) and induces apoptosis only if freed from both. The finding that different prosurvival proteins have selective roles has notable implications for the design of anti-cancer drugs that target the Bcl-2 family.
Figures

References
-
- Adams J.M. 2003. Ways of dying: Multiple pathways to apoptosis. Genes & Dev. 17: 2481-2495. - PubMed
-
- Antonsson B., Montessuit, S., Sanchez, B., and Martinou, J.C. 2001. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 276: 11615-11623. - PubMed
-
- Bae J., Leo, C.P., Hsu, S.Y., and Hsueh, A.H. 2000. Mcl-1S, a splicing variant of the antiapoptotic Bcl-2 family member Mcl-1, encodes a proapoptotic protein possessing only the BH3 domain. J. Biol. Chem. 275: 25255-25261. - PubMed
-
- Chen L., Willis, S.N., Wei, A., Smith, B.J., Fletcher, J.I., Hinds, M.G., Colman, P.M., Day, C.L., Adams, J.M., and Huang, D.C.S. 2005. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17: 393-403. - PubMed
-
- Cheng E.H.-Y., Levine, B., Boise, L.H., Thompson, C.G., and Hardwick, J.M. 1996. Bax-independent inhibition of apoptosis by Bcl-xL. Nature 379: 554-556. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials