

The pattern of polymorphism in Arabidopsis thaliana
- PMID: 15907155
- PMCID: PMC1135296
- DOI: 10.1371/journal.pbio.0030196
The pattern of polymorphism in Arabidopsis thaliana
Abstract
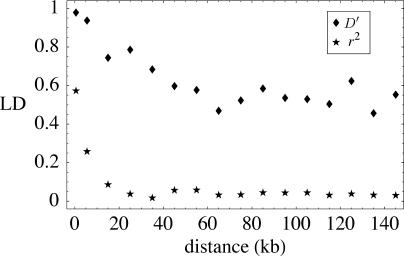
We resequenced 876 short fragments in a sample of 96 individuals of Arabidopsis thaliana that included stock center accessions as well as a hierarchical sample from natural populations. Although A. thaliana is a selfing weed, the pattern of polymorphism in general agrees with what is expected for a widely distributed, sexually reproducing species. Linkage disequilibrium decays rapidly, within 50 kb. Variation is shared worldwide, although population structure and isolation by distance are evident. The data fail to fit standard neutral models in several ways. There is a genome-wide excess of rare alleles, at least partially due to selection. There is too much variation between genomic regions in the level of polymorphism. The local level of polymorphism is negatively correlated with gene density and positively correlated with segmental duplications. Because the data do not fit theoretical null distributions, attempts to infer natural selection from polymorphism data will require genome-wide surveys of polymorphism in order to identify anomalous regions. Despite this, our data support the utility of A. thaliana as a model for evolutionary functional genomics.
Figures

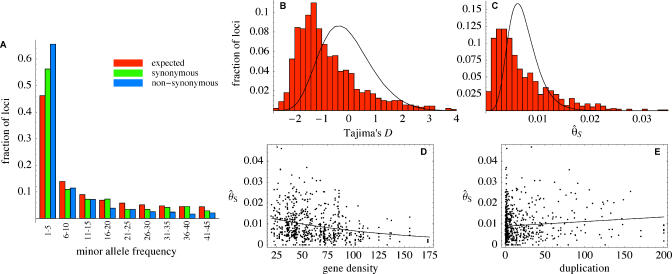
 . The excess of rare alleles is largely limited to frequencies one and two. (B) The distribution of Tajima's D statistic [27] across the sequenced fragments, along with its expected distribution in a constant population (estimated by simulating 1,000 datasets matching the real one in terms of exon/nonexon composition and sample size). (C) The distribution of the level of polymorphism (θ^S
) across the sequenced fragments along with its expected distribution (estimated the same way). (D) The level of polymorphism in nonexon sequences as a function of the local gene density (measured in open reading frames per centimorgan). (E) The level of polymorphism in nonexon sequences as a function of the degree of duplication in each fragment (measured as the negative log10 of the BLAST significance for the second-best hit in the genome). The patterns in (D) and (E) are also seen in exons.
. The excess of rare alleles is largely limited to frequencies one and two. (B) The distribution of Tajima's D statistic [27] across the sequenced fragments, along with its expected distribution in a constant population (estimated by simulating 1,000 datasets matching the real one in terms of exon/nonexon composition and sample size). (C) The distribution of the level of polymorphism (θ^S
) across the sequenced fragments along with its expected distribution (estimated the same way). (D) The level of polymorphism in nonexon sequences as a function of the local gene density (measured in open reading frames per centimorgan). (E) The level of polymorphism in nonexon sequences as a function of the degree of duplication in each fragment (measured as the negative log10 of the BLAST significance for the second-best hit in the genome). The patterns in (D) and (E) are also seen in exons.References
-
- Kreitman M. Nucleotide polymorphism at the alcohol dehydrogenase locus of Drosophila melanogaster . Nature. 1983;304:412–417. - PubMed
-
- Li WH. Molecular evolution. Sunderland (Massachusetts): Sinauer Associates; 1997. 487 pp.
-
- Kreitman M. Methods to detect selection in populations with applications to the human. Annu Rev Genomics Hum Genet. 2000;1:539–559. - PubMed
-
- Stephens M. Inference under the coalescent. In: Balding DJ, Bishop MJ, Cannings C, editors. Handbook of statistical genetics. Chichester (United Kingdom): John Wiley and Sons; 2001. pp. 213–238.
