

An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants
- PMID: 15907219
- PMCID: PMC1173109
- DOI: 10.1186/1471-2180-5-30
An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants
Abstract
Background: Traditional gene replacement procedures are still time-consuming. They usually necessitate cloning of the gene to be mutated, insertional inactivation of the gene with an antibiotic resistance cassette and exchange of the plasmid-borne mutant allele with the bacterial chromosome. PCR and recombinational technologies can be exploited to substantially accelerate virtually all steps involved in the gene replacement process.
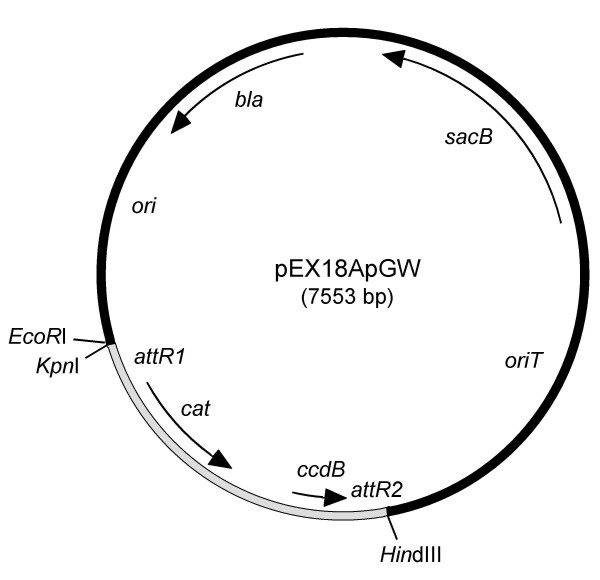
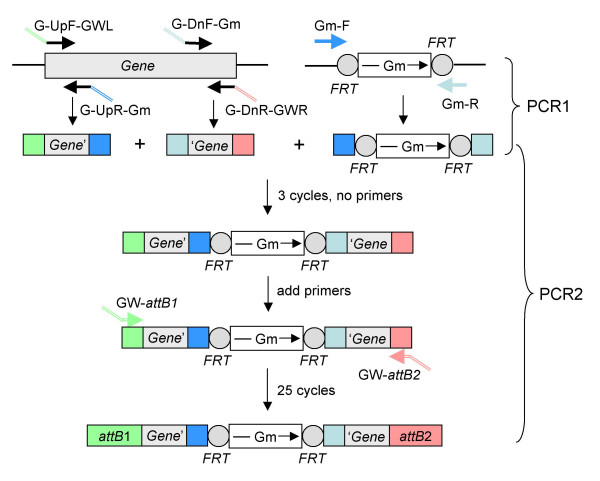
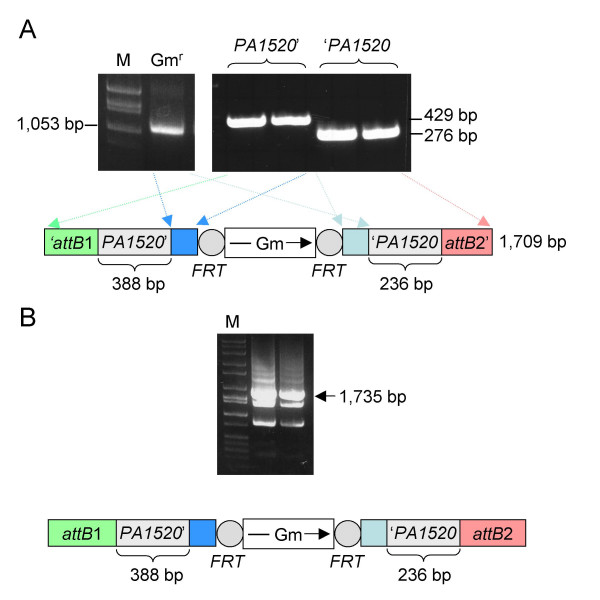
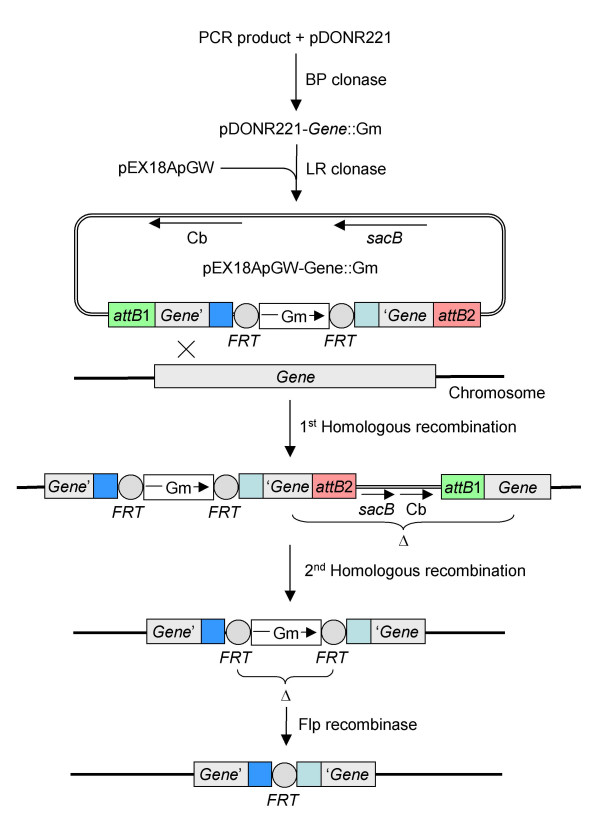
Results: We describe a method for rapid generation of unmarked P. aeruginosa deletion mutants. Three partially overlapping DNA fragments are amplified and then spliced together in vitro by overlap extension PCR. The resulting DNA fragment is cloned in vitro into the Gateway vector pDONR221 and then recombined into the Gateway-compatible gene replacement vector pEX18ApGW. The plasmid-borne deletions are next transferred to the P. aeruginosa chromosome by homologous recombination. Unmarked deletion mutants are finally obtained by Flp-mediated excision of the antibiotic resistance marker. The method was applied to deletion of 25 P. aeruginosa genes encoding transcriptional regulators of the GntR family.
Conclusion: While maintaining the key features of traditional gene replacement procedures, for example, suicide delivery vectors, antibiotic resistance selection and sucrose counterselection, the method described here is considerably faster due to streamlining of some of the key steps involved in the process, especially plasmid-borne mutant allele construction and its transfer into the target host. With appropriate modifications, the method should be applicable to other bacteria.
Figures
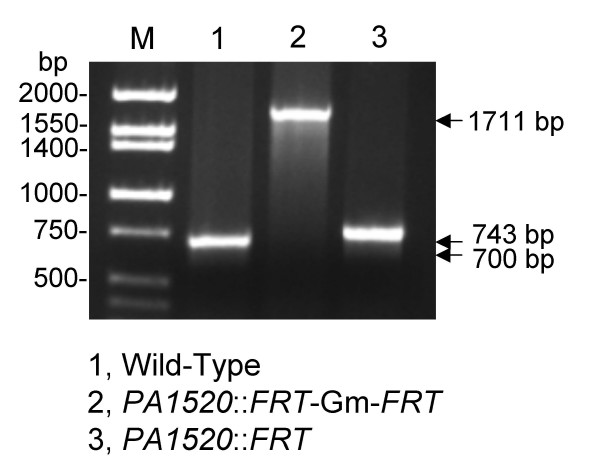
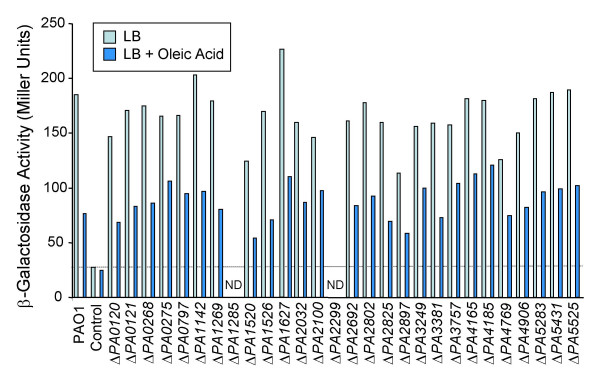
Similar articles
-
A rapid seamless method for gene knockout in Pseudomonas aeruginosa.BMC Microbiol. 2017 Sep 19;17(1):199. doi: 10.1186/s12866-017-1112-5. BMC Microbiol. 2017. PMID: 28927382 Free PMC article.
-
Suicide vectors for antibiotic marker exchange and rapid generation of multiple knockout mutants by allelic exchange in Gram-negative bacteria.J Microbiol Methods. 2006 Dec;67(3):395-407. doi: 10.1016/j.mimet.2006.04.011. Epub 2006 Jun 5. J Microbiol Methods. 2006. PMID: 16750581
-
A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants.Gene. 1998 May 28;212(1):77-86. doi: 10.1016/s0378-1119(98)00130-9. Gene. 1998. PMID: 9661666
-
Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange.Nat Protoc. 2015 Nov;10(11):1820-41. doi: 10.1038/nprot.2015.115. Epub 2015 Oct 22. Nat Protoc. 2015. PMID: 26492139 Free PMC article.
-
High-Frequency Targeted Mutagenesis in Pseudomonas stutzeri Using a Vector-Free Allele-Exchange Protocol.J Microbiol Biotechnol. 2017 Feb 28;27(2):335-341. doi: 10.4014/jmb.1608.08019. J Microbiol Biotechnol. 2017. PMID: 27817190
Cited by
-
Accumulation of Pyrimidine Intermediate Orotate Decreases Virulence Factor Production in Pseudomonas aeruginosa.Curr Microbiol. 2015 Aug;71(2):229-34. doi: 10.1007/s00284-015-0826-6. Epub 2015 Apr 28. Curr Microbiol. 2015. PMID: 25917504
-
The exopolysaccharide gene cluster Bcam1330-Bcam1341 is involved in Burkholderia cenocepacia biofilm formation, and its expression is regulated by c-di-GMP and Bcam1349.Microbiologyopen. 2013 Feb;2(1):105-22. doi: 10.1002/mbo3.61. Epub 2012 Dec 25. Microbiologyopen. 2013. PMID: 23281338 Free PMC article.
-
A Broad Spectrum Racemase in Pseudomonas putida KT2440 Plays a Key Role in Amino Acid Catabolism.Front Microbiol. 2018 Jun 29;9:1343. doi: 10.3389/fmicb.2018.01343. eCollection 2018. Front Microbiol. 2018. PMID: 30008699 Free PMC article.
-
Transcriptional regulation of fatty acid cis-trans isomerization in the solvent-tolerant soil bacterium, Pseudomonas putida F1.Environ Microbiol. 2019 May;21(5):1659-1676. doi: 10.1111/1462-2920.14546. Epub 2019 Mar 12. Environ Microbiol. 2019. PMID: 30702193 Free PMC article.
-
Small RNA ArrF regulates the expression of sodB and feSII genes in Azotobacter vinelandii.Curr Microbiol. 2008 Dec;57(6):593-7. doi: 10.1007/s00284-008-9248-z. Epub 2008 Sep 25. Curr Microbiol. 2008. PMID: 18830664
References
-
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Spencer D, Wong GKS, Wu Z, Paulsen IT, Reizer J, M.H. Saier. R.E.W. Hancock. Lory S, Olson MV. Complete genome sequence of Pseudomonas aeruginosa, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. - DOI - PubMed
-
- Jacobs MA, Alwood A, Thaipisuttikul I, Spencer DH, Haugen E, Ernst S, Will O, Kaul R, Raymond CK, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. Comprehensive transposon mutant library of Pseudomonas aeruginosa. PNAS. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
