

Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin
- PMID: 15923638
- PMCID: PMC1140594
- DOI: 10.1128/MCB.25.12.5242-5252.2005
Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin
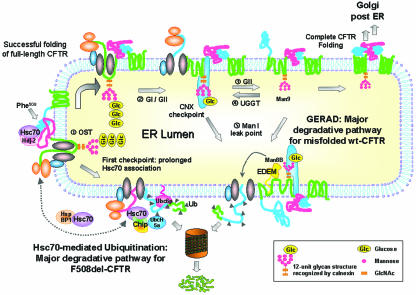
Abstract
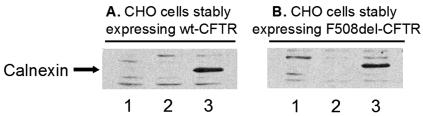
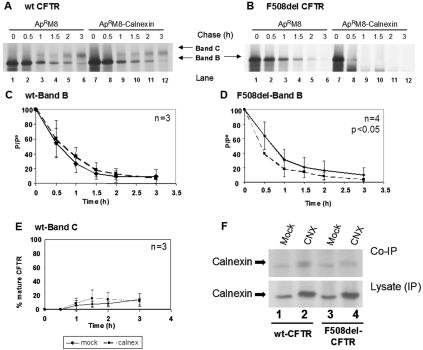
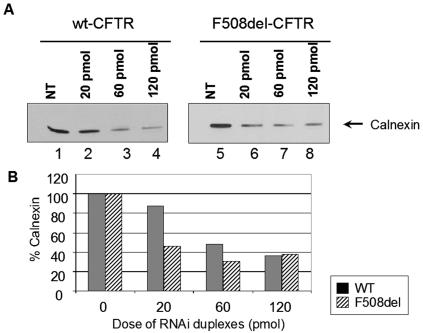
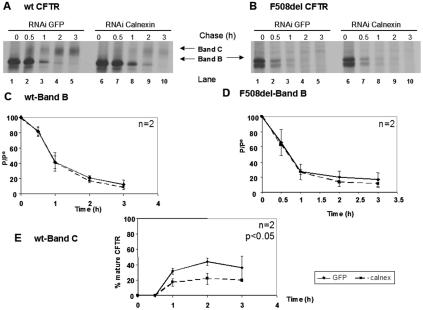
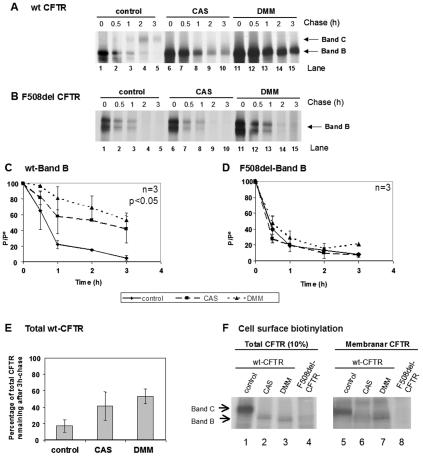
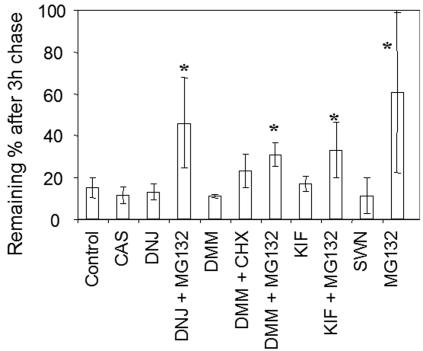
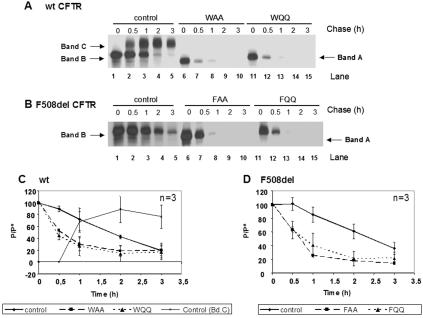
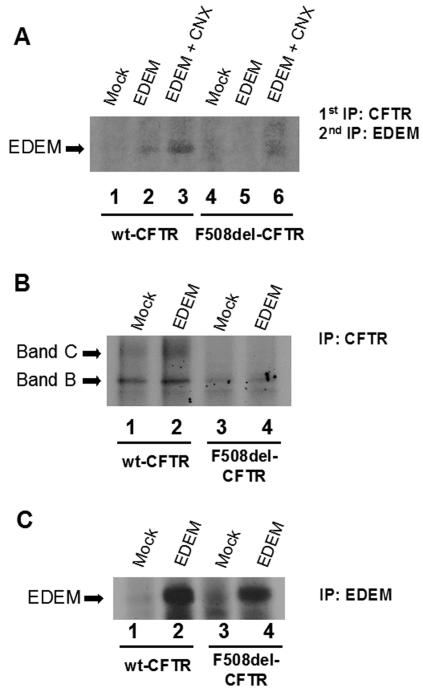
Biosynthesis and folding of multidomain transmembrane proteins is a complex process. Structural fidelity is monitored by endoplasmic reticulum (ER) quality control involving the molecular chaperone calnexin. Retained misfolded proteins undergo ER-associated degradation (ERAD) through the ubiquitin-proteasome pathway. Our data show that the major degradation pathway of the cystic fibrosis transmembrane conductance regulator (CFTR) with F508del (the most frequent mutation found in patients with the genetic disease cystic fibrosis) from the ER is independent of calnexin. Moreover, our results demonstrate that inhibition of mannose-processing enzymes, unlike most substrate glycoproteins, does not stabilize F508del-CFTR, although wild-type (wt) CFTR is drastically stabilized under the same conditions. Together, our data support a novel model by which wt and F508del-CFTR undergo ERAD from two distinct checkpoints, the mutant being disposed of independently of N-glycosidic residues and calnexin, probably by the Hsc70/Hsp70 machinery, and wt CFTR undergoing glycan-mediated ERAD.
Figures
References
-
- Ayalon-Soffer, M., M. Shenkman, and G. Z. Lederkremer. 1999. Differential role of mannose and glucose trimming in the ER degradation of asialoglycoprotein receptor subunits. J. Cell Sci. 112:3309-3318. - PubMed
-
- Bergeron, J. J., M. B. Brenner, D. Y. Thomas, and D. B. Williams. 1994. Calnexin: a membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 19:124-128. - PubMed
-
- Cabral, C. M., Y. Liu, and R. N. Sifers. 2001. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem. Sci. 26:619-624. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous