

The coordinate regulation of the p53 and mTOR pathways in cells
- PMID: 15928081
- PMCID: PMC1142118
- DOI: 10.1073/pnas.0502857102
The coordinate regulation of the p53 and mTOR pathways in cells
Abstract
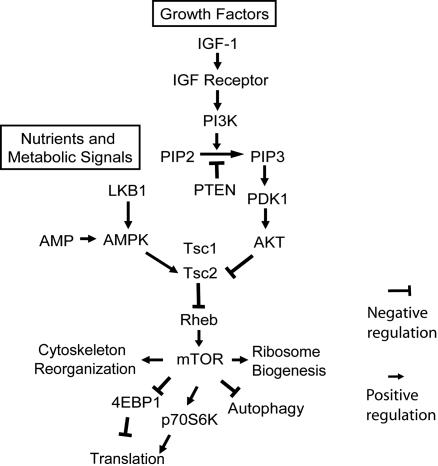
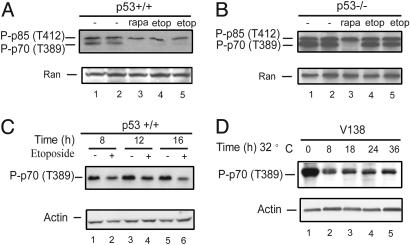
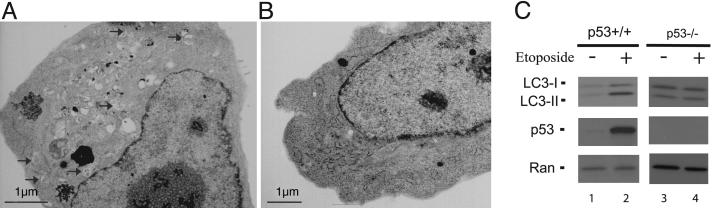
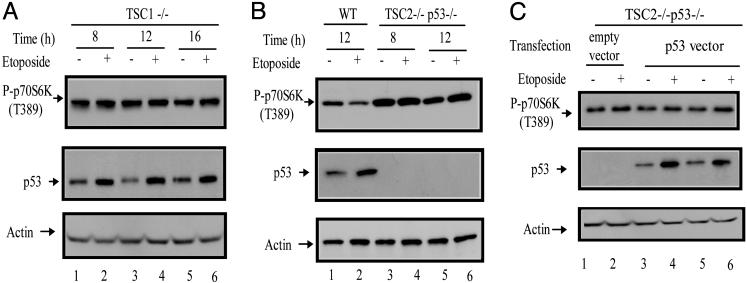
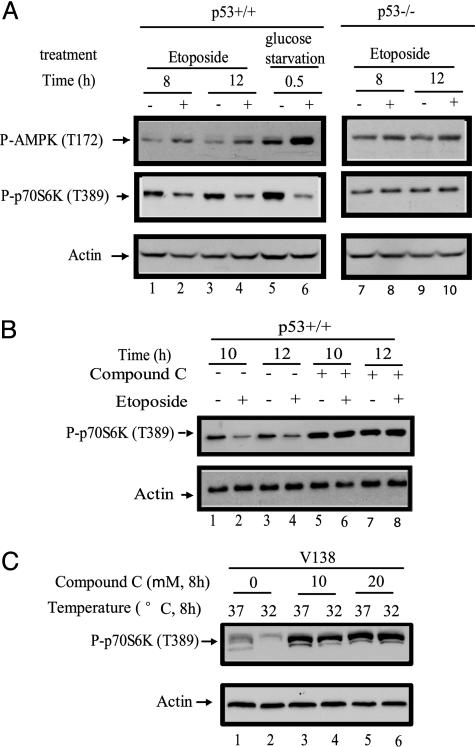
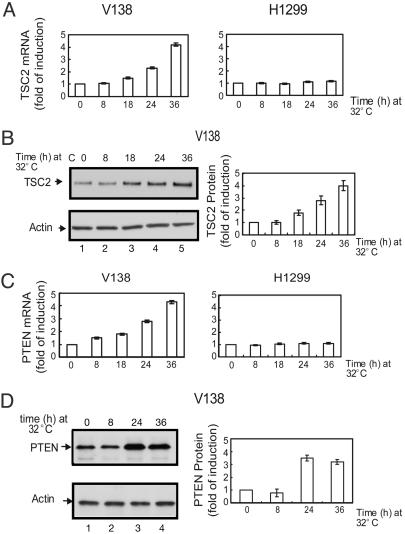
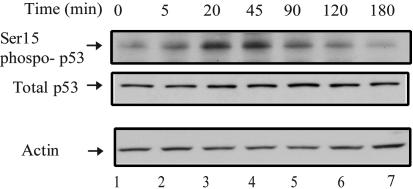
Cell growth and proliferation requires an intricate coordination between the stimulatory signals arising from nutrients and growth factors and the inhibitory signals arising from intracellular and extracellular stresses. Alteration of the coordination often causes cancer. In mammals, the mTOR (mammalian target of rapamycin) protein kinase is the central node in nutrient and growth factor signaling, and p53 plays a critical role in sensing genotoxic and other stresses. The results presented here demonstrate that activation of p53 inhibits mTOR activity and regulates its downstream targets, including autophagy, a tumor suppression process. Moreover, the mechanisms by which p53 regulates mTOR involves AMP kinase activation and requires the tuberous sclerosis (TSC) 1/TSC2 complex, both of which respond to energy deprivation in cells. In addition, glucose starvation not only signals to shut down mTOR, but also results in the transient phosphorylation of the p53 protein. Thus, p53 and mTOR signaling machineries can cross-talk and coordinately regulate cell growth, proliferation, and death.
Figures
References
-
- Hay, N. & Sonenberg, N. (2004) Genes Dev. 18, 1926-1945. - PubMed
-
- Harris, T. E. & Lawrence, J. C., Jr. (2003) Sci. STKE 2003, re15. - PubMed
-
- Kim, D. H. & Sabatini, D. M. (2004) Curr. Top. Microbiol. Immunol. 279, 259-270. - PubMed
-
- Inoki, K., Zhu, T. & Guan, K. L. (2003) Cell 115, 577-590. - PubMed
-
- Brown, E. J., Beal, P. A., Keith, C. T., Chen, J., Shin, T. B. & Schreiber, S. L. (1995) Nature 377, 441-446. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
