

SplitTester: software to identify domains responsible for functional divergence in protein family
- PMID: 15929795
- PMCID: PMC1181622
- DOI: 10.1186/1471-2105-6-137
SplitTester: software to identify domains responsible for functional divergence in protein family
Abstract
Background: Many protein families have undergone functional divergence after gene duplications such that current subgroups of the family carry out overlapping but distinct biological roles. For the protein families with known functional subtypes (a functional split), we developed the software, SplitTester, to identify potential regions that are responsible for the observed distinct functional subtypes within the same protein family.
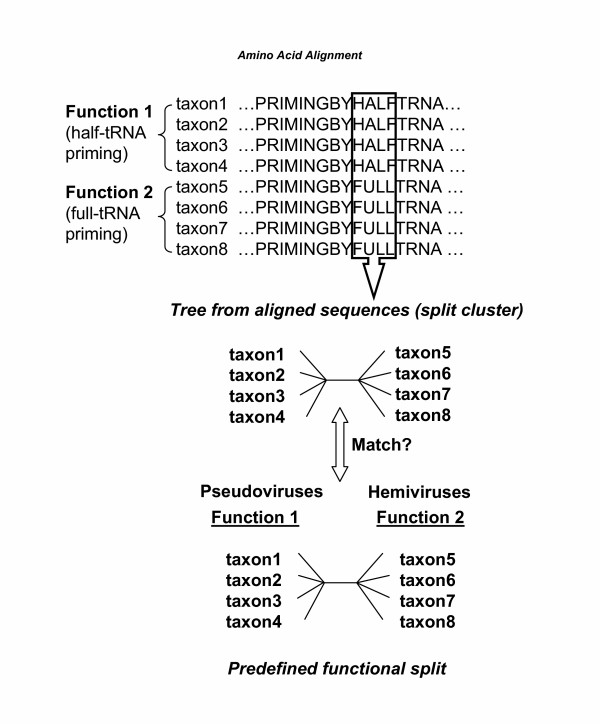
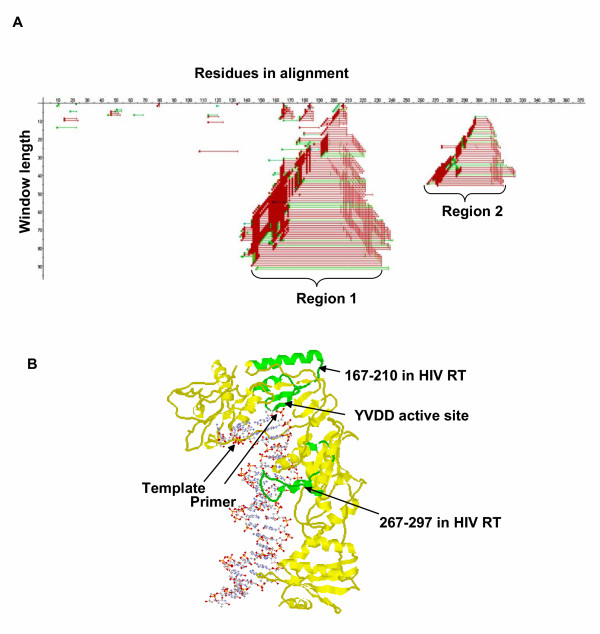
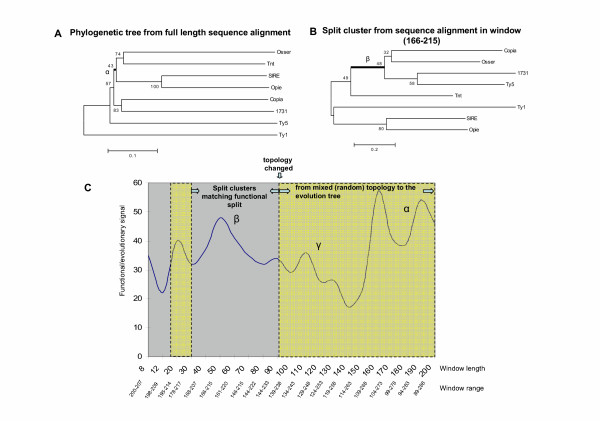
Results: Our software, SplitTester, takes a multiple protein sequences alignment as input, generated from protein members of two subgroups with known functional divergence. SplitTester was designed to construct the neighbor joining tree (a split cluster) from variable-sized sliding windows across the alignment in a process called split-clustering. SplitTester identifies the regions, whose split cluster is consistent with the functional split, but may be inconsistent with the phylogeny of the protein family. We hypothesize that at least some number of these identified regions, which are not following a random mutation process, are responsible for the observed functional split. To test our method, we used reverse transcriptase from a group of Pseudoviridae retrotransposons: to identify residues specific for diverged primer recognition. Candidate regions were then mapped onto the three dimensional structures of reverse transcriptase. The locations of these amino acids within the enzyme are consistent with their biological roles.
Conclusion: SplitTester aims to identify specific domain sequences responsible for functional divergence of subgroups within a protein family. From the analysis of retroelements reverse transcriptase family, we successfully identified the regions splitting this family according to the primer specificity, implying their functions in the specific primer selection.
Figures

Similar articles
-
BADASP: predicting functional specificity in protein families using ancestral sequences.Bioinformatics. 2005 Nov 15;21(22):4190-1. doi: 10.1093/bioinformatics/bti678. Epub 2005 Sep 13. Bioinformatics. 2005. PMID: 16159912
-
DIVERGE: phylogeny-based analysis for functional-structural divergence of a protein family.Bioinformatics. 2002 Mar;18(3):500-1. doi: 10.1093/bioinformatics/18.3.500. Bioinformatics. 2002. PMID: 11934757
-
CAPS: coevolution analysis using protein sequences.Bioinformatics. 2006 Nov 15;22(22):2821-2. doi: 10.1093/bioinformatics/btl493. Epub 2006 Sep 27. Bioinformatics. 2006. PMID: 17005535
-
[From sequence variability to structural and functional prediction: modeling of homologous protein families].Biol Aujourdhui. 2017;211(3):239-244. doi: 10.1051/jbio/2017030. Epub 2018 Feb 7. Biol Aujourdhui. 2017. PMID: 29412135 Review. French.
-
Automated detection of remote homology.Curr Opin Struct Biol. 2002 Jun;12(3):362-7. doi: 10.1016/s0959-440x(02)00332-9. Curr Opin Struct Biol. 2002. PMID: 12127456 Review.
Cited by
-
Divergent evolution of the chloroplast small heat shock protein gene in the genera Rhododendron (Ericaceae) and Machilus (Lauraceae).Ann Bot. 2007 Mar;99(3):461-75. doi: 10.1093/aob/mcl288. Epub 2007 Feb 9. Ann Bot. 2007. PMID: 17293350 Free PMC article.
-
Genome-wide functional divergence after the symbiosis of proteobacteria with insects unraveled through a novel computational approach.PLoS Comput Biol. 2009 Apr;5(4):e1000344. doi: 10.1371/journal.pcbi.1000344. Epub 2009 Apr 3. PLoS Comput Biol. 2009. PMID: 19343224 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
