

Discovering regulatory binding-site modules using rule-based learning
- PMID: 15930496
- PMCID: PMC1142476
- DOI: 10.1101/gr.3760605
Discovering regulatory binding-site modules using rule-based learning
Abstract
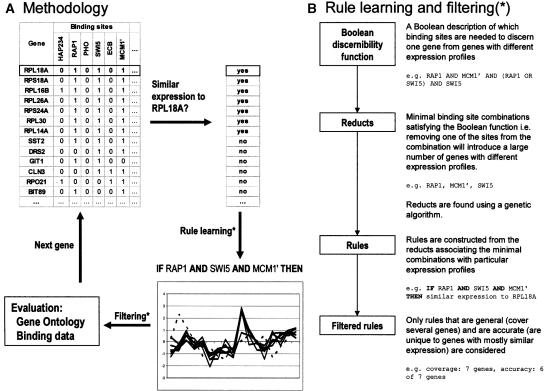
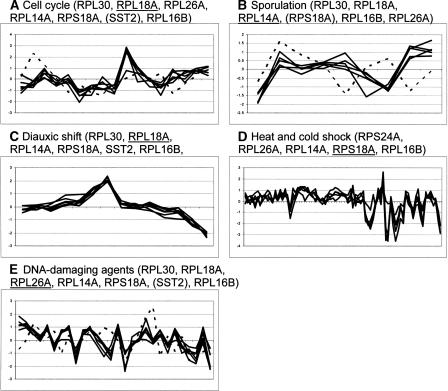
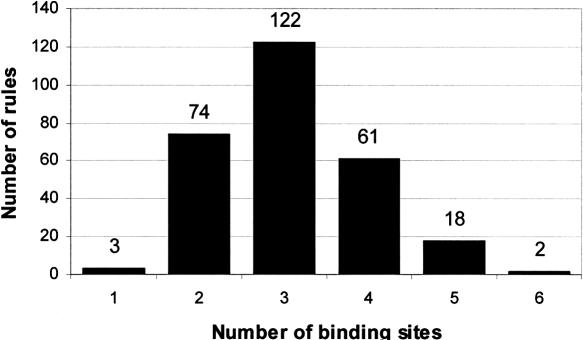
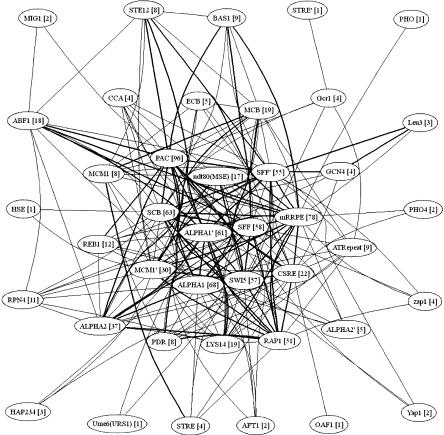
Transcription factors regulate expression by binding selectively to sequence sites in cis-regulatory regions of genes. It is therefore reasonable to assume that genes regulated by the same transcription factors should all contain the corresponding binding sites in their regulatory regions and exhibit similar expression profiles as measured by, for example, microarray technology. We have used this assumption to analyze genome-wide yeast binding-site and microarray expression data to reveal the combinatorial nature of gene regulation. We obtained IF-THEN rules linking binding-site combinations (binding-site modules) to genes with particular expression profiles, and thereby provided testable hypotheses on the combinatorial coregulation of gene expression. We showed that genes associated with such rules have a significantly higher probability of being bound by the same transcription factors, as indicated by a genome-wide location analysis, than genes associated with only common binding sites or similar expression. Furthermore, we also found that such genes were significantly more often biologically related in terms of Gene Ontology annotations than genes only associated with common binding sites or similar expression. We analyzed expression data collected under different sets of stress conditions and found many binding-site modules that are conserved over several of these condition sets, as well as modules that are specific to particular biological responses. Our results on the reoccurrence of binding sites in different modules provide specific data on how binding sites may be combined to allow a large number of expression outcomes using relatively few transcription factors.
Figures
References
-
- Aach, J., Rindone, W., and Church, G.M. 2000. Systematic management and analysis of yeast gene expression data. Genome Res. 10: 431-445. - PubMed
-
- Beer, M.A. and Tavazoie, S. 2004. Predicting gene expression from sequence. Cell 117: 185-198. - PubMed
-
- Berman, B.P., Nibu, Y., Pfeiffer, B.D., Tomancak, P., Celniker, S.E., Levine, M., Rubin, G.M., and Eisen, M.B. 2002. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl. Acad. Sci. 99: 757-762. - PMC - PubMed
WEB SITE REFERENCES
-
- http://salt2.med.harvard.edu/ExpressDB/; ExpressDB, database for gene expression data.
-
- http://genetics.med.harvard.edu/~tpilpel/MotComb.html; Web supplement to Pilpel et al. (2001).
-
- http://rosetta.lcb.uu.se; the ROSETTA system.
-
- http://www.geneontology.org; Gene Ontology.
-
- http://www.lcb.uu.se/~hvidsten/binding_sites/; our Web site with the Supplemental Material.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases