

Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir
- PMID: 15937277
- PMCID: PMC2253353
- DOI: 10.1110/ps.051347405
Structural analysis of an HIV-1 protease I47A mutant resistant to the protease inhibitor lopinavir
Abstract
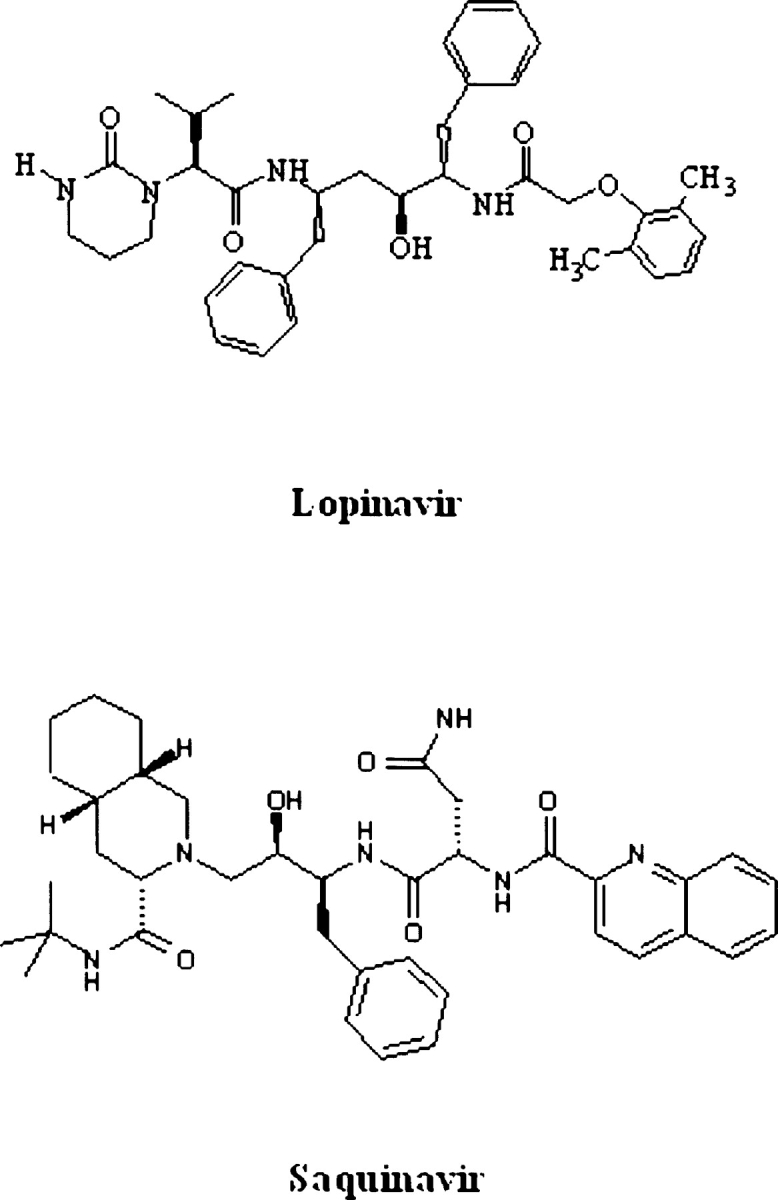
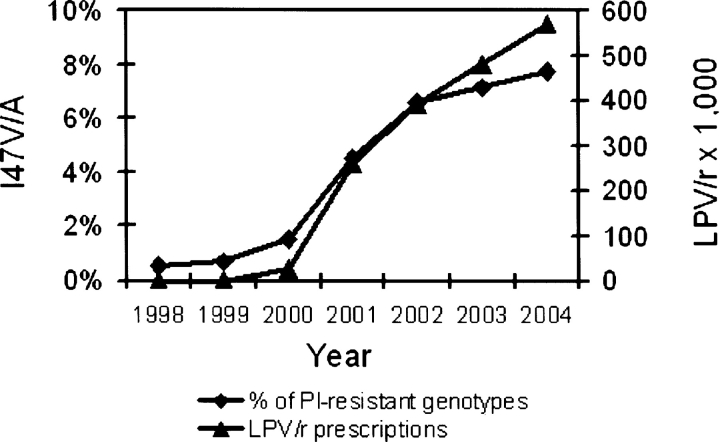
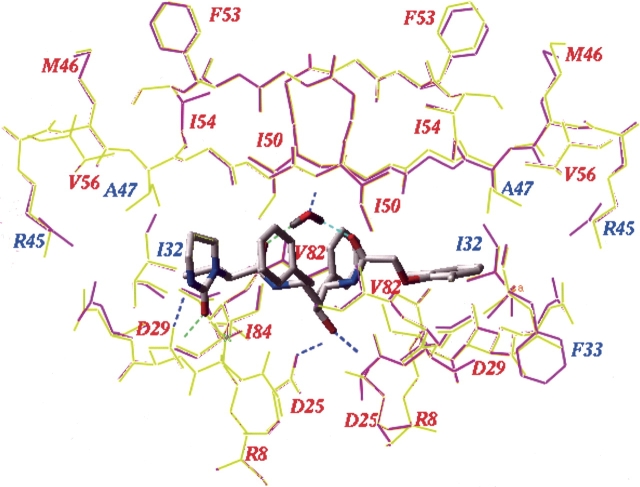
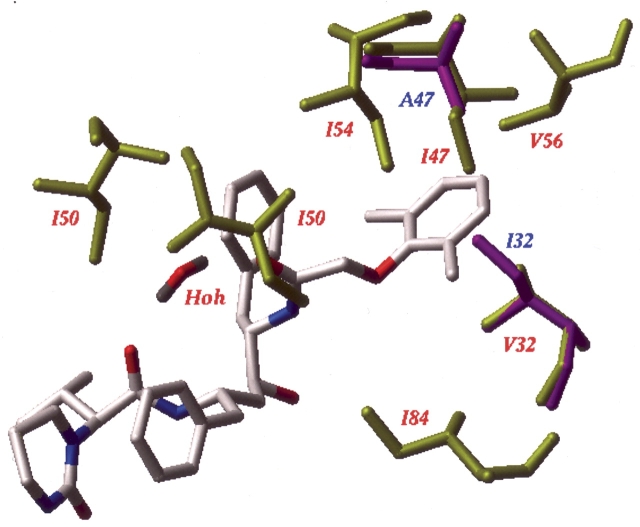
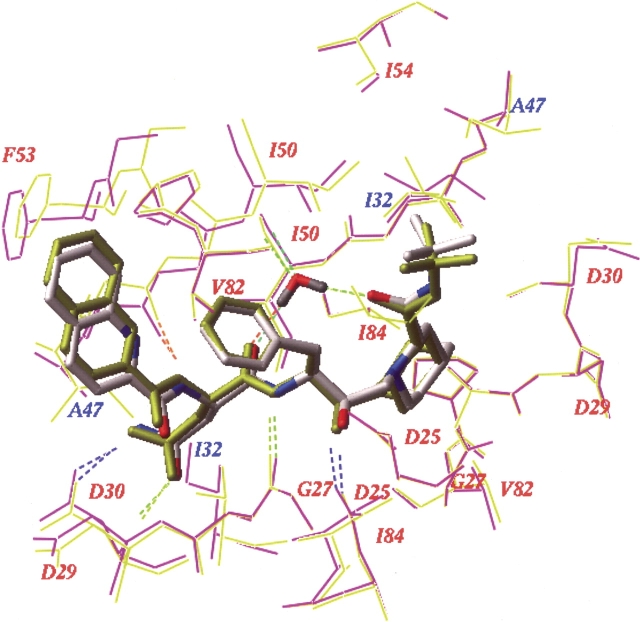
We have identified a rare HIV-1 protease (PR) mutation, I47A, associated with a high level of resistance to the protease inhibitor lopinavir (LPV) and with hypersusceptibility to the protease inhibitor saquinavir (SQV). The I47A mutation was found in 99 of 112,198 clinical specimens genotyped after LPV became available in late 2000, but in none of 24,426 clinical samples genotyped from 1998 to October 2000. Phenotypic data obtained for five I47A mutants showed unexpected resistance to LPV (86- to >110-fold) and hypersusceptibility to SQV (0.1- to 0.7-fold). Molecular modeling and energy calculations for these mutants using our structural phenotyping methodology showed an increase in the binding energy of LPV by 1.9-3.1 kcal/mol with respect to the wild type complex, corresponding to a 20- to >100-fold decrease in binding affinity, consistent with the observed high levels of LPV resistance. In the WT PR-LPV complex, the Ile 47 side chain is positioned close to the phenoxyacetyl moiety of LPV and its van der Waals interactions contribute significantly to the ligand binding. These interactions are lost for the smaller Ala 47 residue. Calculated binding energy changes for SQV ranged from -0.4 to -1.2 kcal/mol. In the mutant I47A PR-SQV complexes, the PR flaps are packed more tightly around SQV than in the WT complex, resulting in the formation of additional hydrogen bonds that increase binding affinity of SQV consistent with phenotypic hypersusceptibility. The emergence of mutations at PR residue 47 strongly correlates with increasing prescriptions of LPV (Spearman correlation r(s) = 0.96, P < .0001).
Figures
References
-
- Abagyan, R. and Totrov, M. 1994. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 235 983–1002. - PubMed
-
- Abagyan, R., Totrov, M., and Kuznetsov, D. 1994. ICM A new method for protein modeling and design: Applications to docking and structure prediction from the disorded red native conformation. J. Comput. Chem. 15 488–506.
-
- Baxter, J.D., Mayers, D.L., Wentworth, D.N., Neaton, J.D., Hoover, M.L., Winters, M.A., Mannheimer, S.B., Thompson, M.A., Abrams, D.I., Brizz, B.J., et al. 2000. A randomized study of antiretroviral management based on plasma genotypic antiretroviral resistance testing in patients failing therapy. CPCRA 046 Study Team for the Terry Beirn Community Programs for Clinical Research on AIDS. AIDS 14 F83–93. - PubMed
-
- Benson, C.A., Deeks, S.G., Brun, S.C., Gulick, R.M., Eron, J.J., Kessler, H.A., Murphy, R.L., Hicks, C., King, M., Wheeler, D., et al. 2002. Safety and antiviral activity at 48 weeks of lopinavir/ritonavir plus nevirapine and 2 nucleoside reverse-transcriptase inhibitors in human immunodeficiency virus type 1-infected protease inhibitor-experienced patients. J. Infect. Dis. 185 599–607. - PubMed
-
- Cao, Z.H., Han, L.Y., Zheng, C.J., Lang, Z., Chen, J.X., Lin, H.H., and Chen, Y.Z. 2005. Computer prediction of drug resistance mutations in proteins. Drug Discov. Today 10 521–529. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
