

DOM-fold: a structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain
- PMID: 15937278
- PMCID: PMC2253344
- DOI: 10.1110/ps.051364905
DOM-fold: a structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain
Abstract
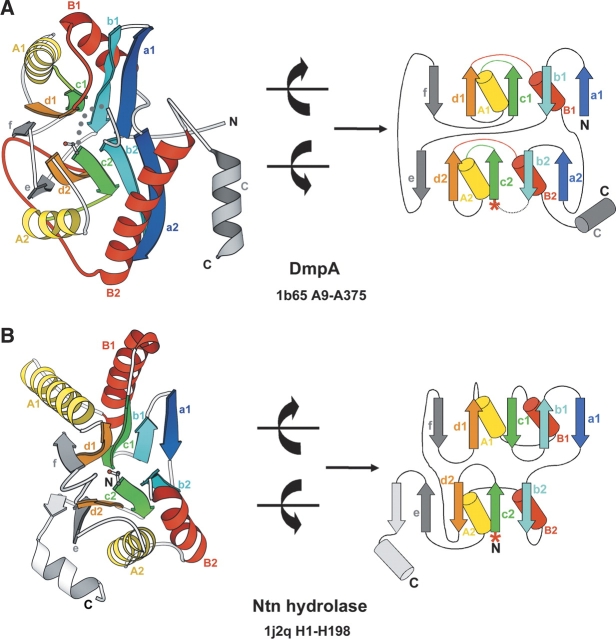
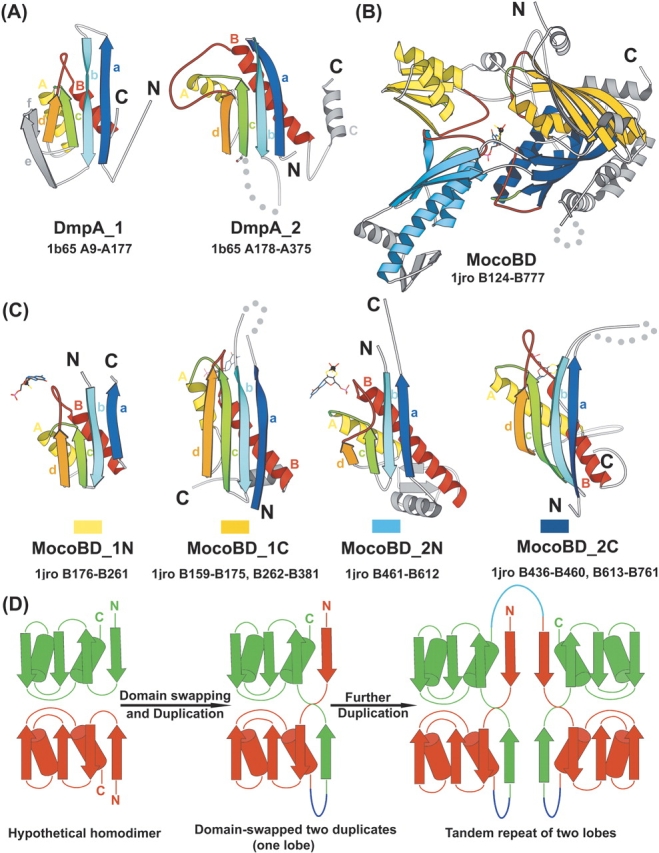
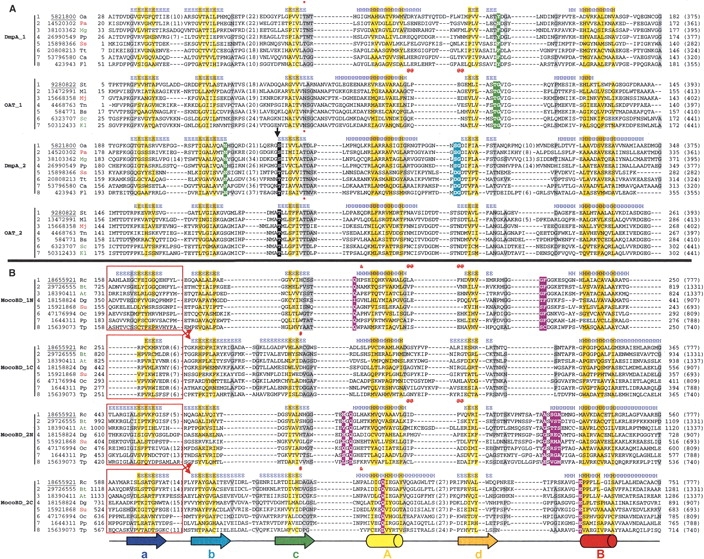
Understanding relationships between sequence, structure, and evolution is important for functional characterization of proteins. Here, we define a novel DOM-fold as a consensus structure of the domains in DmpA (L-aminopeptidase D-Ala-esterase/amidase), OAT (ornithine acetyltransferase), and MocoBD (molybdenum cofactor-binding domain), and discuss possible evolutionary scenarios of its origin. As shown by a comprehensive structure similarity search, DOM-fold distinguished by a two-layered beta/alpha architecture of a particular topology with unusual crossing loops is unique to those three protein families. DmpA and OAT are evolutionarily related as indicated by their sequence, structural, and functional similarities. Structural similarity between the DmpA/OAT superfamily and the MocoBD domains has not been reported before. Contrary to previous reports, we conclude that functional similarities between DmpA/OAT proteins and N-terminal nucleophile (Ntn) hydrolases are convergent and are unlikely to be inherited from a common ancestor.
Figures
Similar articles
-
A new variant of the Ntn hydrolase fold revealed by the crystal structure of L-aminopeptidase D-ala-esterase/amidase from Ochrobactrum anthropi.Structure. 2000 Feb 15;8(2):153-62. doi: 10.1016/s0969-2126(00)00091-5. Structure. 2000. PMID: 10673442
-
X-ray crystal structure of ornithine acetyltransferase from the clavulanic acid biosynthesis gene cluster.Biochem J. 2005 Jan 15;385(Pt 2):565-73. doi: 10.1042/BJ20040814. Biochem J. 2005. PMID: 15352873 Free PMC article.
-
Insights into molybdenum cofactor deficiency provided by the crystal structure of the molybdenum cofactor biosynthesis protein MoaC.Structure. 2000 Jul 15;8(7):709-18. doi: 10.1016/s0969-2126(00)00157-x. Structure. 2000. PMID: 10903949
-
Enzymes depending on the pterin molybdenum cofactor: sequence families, spectroscopic properties of molybdenum and possible cofactor-binding domains.Biochim Biophys Acta. 1991 Mar 29;1057(2):157-85. doi: 10.1016/s0005-2728(05)80100-8. Biochim Biophys Acta. 1991. PMID: 2015248 Review. No abstract available.
-
Molybdenum and tungsten oxygen transferases--and functional diversity within a common active site motif.Metallomics. 2014 Jan;6(1):15-24. doi: 10.1039/c3mt00177f. Metallomics. 2014. PMID: 24068390 Review.
Cited by
-
Divergence and convergence in enzyme evolution.J Biol Chem. 2012 Jan 2;287(1):21-28. doi: 10.1074/jbc.R111.241976. Epub 2011 Nov 8. J Biol Chem. 2012. PMID: 22069324 Free PMC article. Review.
-
Insights into cis-autoproteolysis reveal a reactive state formed through conformational rearrangement.Proc Natl Acad Sci U S A. 2012 Feb 14;109(7):2308-13. doi: 10.1073/pnas.1113633109. Epub 2012 Jan 30. Proc Natl Acad Sci U S A. 2012. PMID: 22308359 Free PMC article.
-
Three-dimensional structure of nylon hydrolase and mechanism of nylon-6 hydrolysis.J Biol Chem. 2012 Feb 10;287(7):5079-90. doi: 10.1074/jbc.M111.321992. Epub 2011 Dec 19. J Biol Chem. 2012. PMID: 22187439 Free PMC article.
-
β-Aminopeptidases: Insight into Enzymes without a Known Natural Substrate.Appl Environ Microbiol. 2019 Jul 18;85(15):e00318-19. doi: 10.1128/AEM.00318-19. Print 2019 Aug 1. Appl Environ Microbiol. 2019. PMID: 31126950 Free PMC article.
-
Exploring the role of conformational heterogeneity in cis-autoproteolytic activation of ThnT.Biochemistry. 2014 Jul 8;53(26):4273-81. doi: 10.1021/bi500385d. Epub 2014 Jun 26. Biochemistry. 2014. PMID: 24933323 Free PMC article.
References
-
- Abadjieva, A., Hilven, P., Pauwels, K., and Crabeel, M. 2000. The yeast ARG7 gene product is autoproteolyzed to two subunit peptides, yielding active ornithine acetyltransferase. J. Biol. Chem. 275 11361–11367. - PubMed
-
- Bompard-Gilles, C., Villeret, V., Davies, G.J., Fanuel, L., Joris, B., Frere, J.M., and Van Beeumen, J. 2000. A new variant of the Ntn hydrolase fold revealed by the crystal structure of L-aminopeptidase D-ala-esterase/amidase from Ochrobactrum anthropi. Struct. Fold Des. 8 153–162. - PubMed
-
- Bonin, I., Martins, B.M., Purvanov, V., Fetzner, S., Huber, R., and Dobbek, H. 2004. Active site geometry and substrate recognition of the molybdenum hydroxylase quinoline 2-oxidoreductase. Structure (Camb.) 12 1425–1435. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
