

Divergent and convergent evolution after a common-source outbreak of hepatitis C virus
- PMID: 15939791
- PMCID: PMC2213258
- DOI: 10.1084/jem.20050122
Divergent and convergent evolution after a common-source outbreak of hepatitis C virus
Abstract
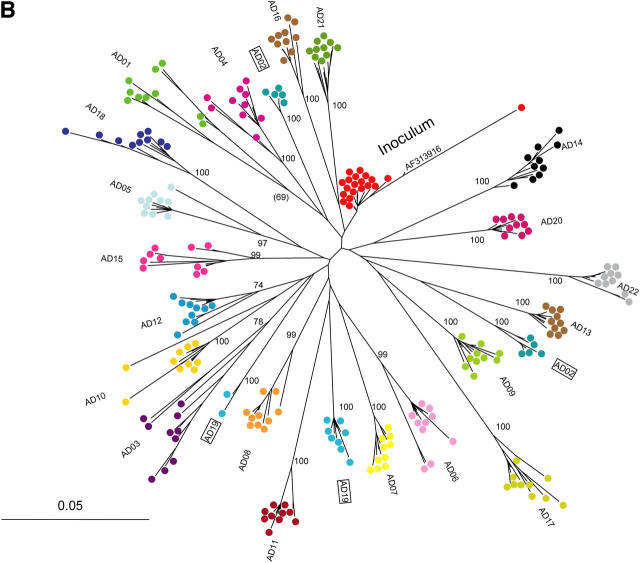
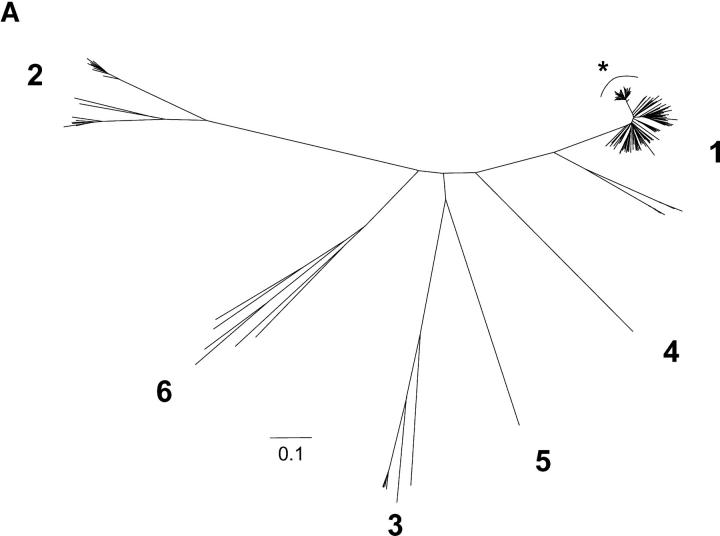
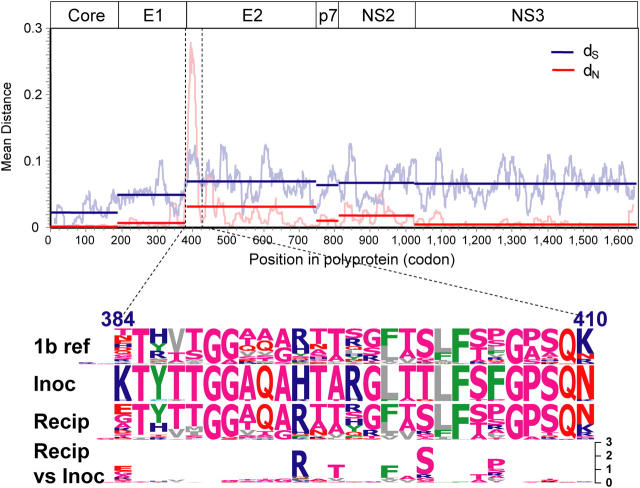
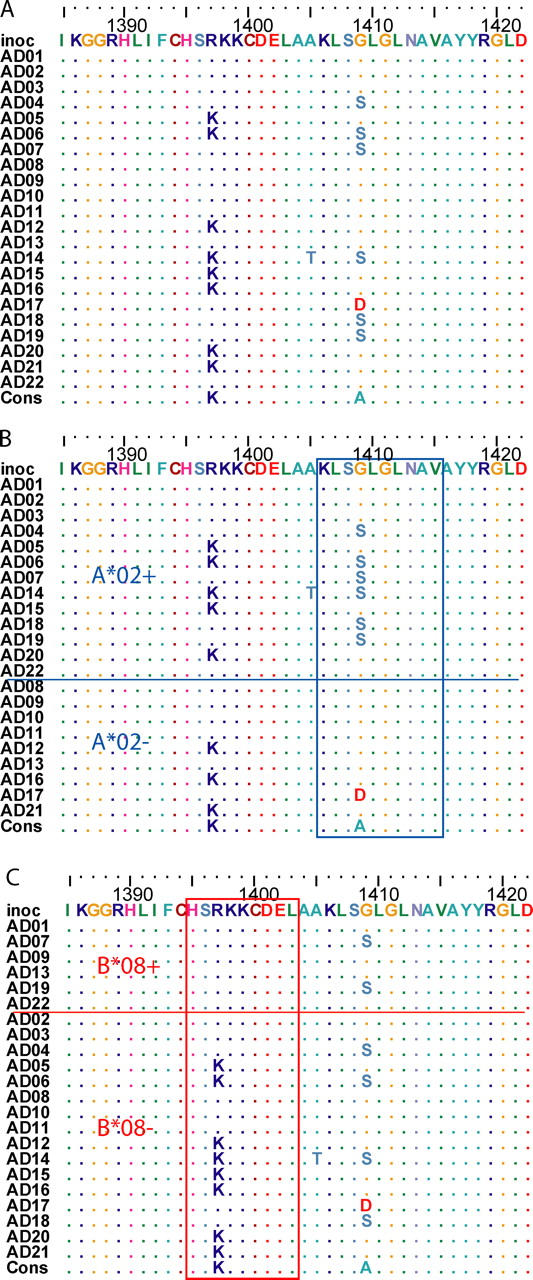
The genomic sequences of viruses that are highly mutable and cause chronic infection tend to diverge over time. We report that these changes represent both immune-driven selection and, in the absence of immune pressure, reversion toward an ancestral consensus. Sequence changes in hepatitis C virus (HCV) structural and nonstructural genes were studied in a cohort of women accidentally infected with HCV in a rare common-source outbreak. We compared sequences present in serum obtained 18-22 yr after infection to sequences present in the shared inoculum and found that HCV evolved along a distinct path in each woman. Amino acid substitutions in known epitopes were directed away from consensus in persons having the HLA allele associated with that epitope (immune selection), and toward consensus in those lacking the allele (reversion). These data suggest that vaccines for genetically diverse viruses may be more effective if they represent consensus sequence, rather than a human isolate.
Figures
References
-
- Neumann, A.U., N.P. Lam, H. Dahari, D.R. Gretch, T.E. Wiley, T.J. Layden, and A.S. Perelson. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 282:103–107. - PubMed
-
- Gaud, U., B. Langer, T. Petropoulou, H.C. Thomas, and P. Karayiannis. 2003. Changes in hypervariable region 1 of the envelope 2 glycoprotein of hepatitis C virus in children and adults with humoral immune defects. J. Med. Virol. 69:350–356. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
