

Mimicry of a constitutively active pre-B cell receptor in acute lymphoblastic leukemia cells
- PMID: 15939795
- PMCID: PMC2213268
- DOI: 10.1084/jem.20042101
Mimicry of a constitutively active pre-B cell receptor in acute lymphoblastic leukemia cells
Abstract
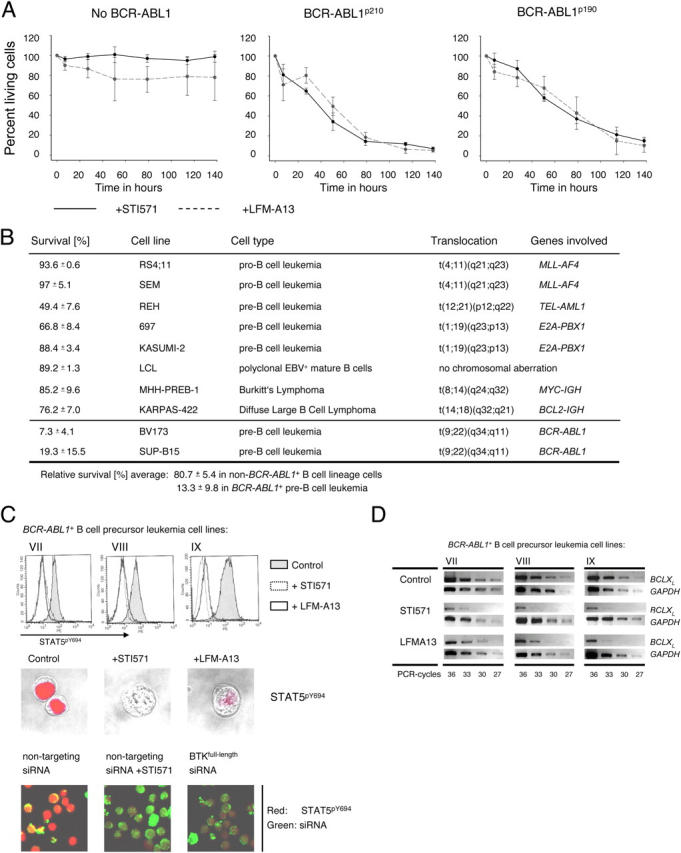
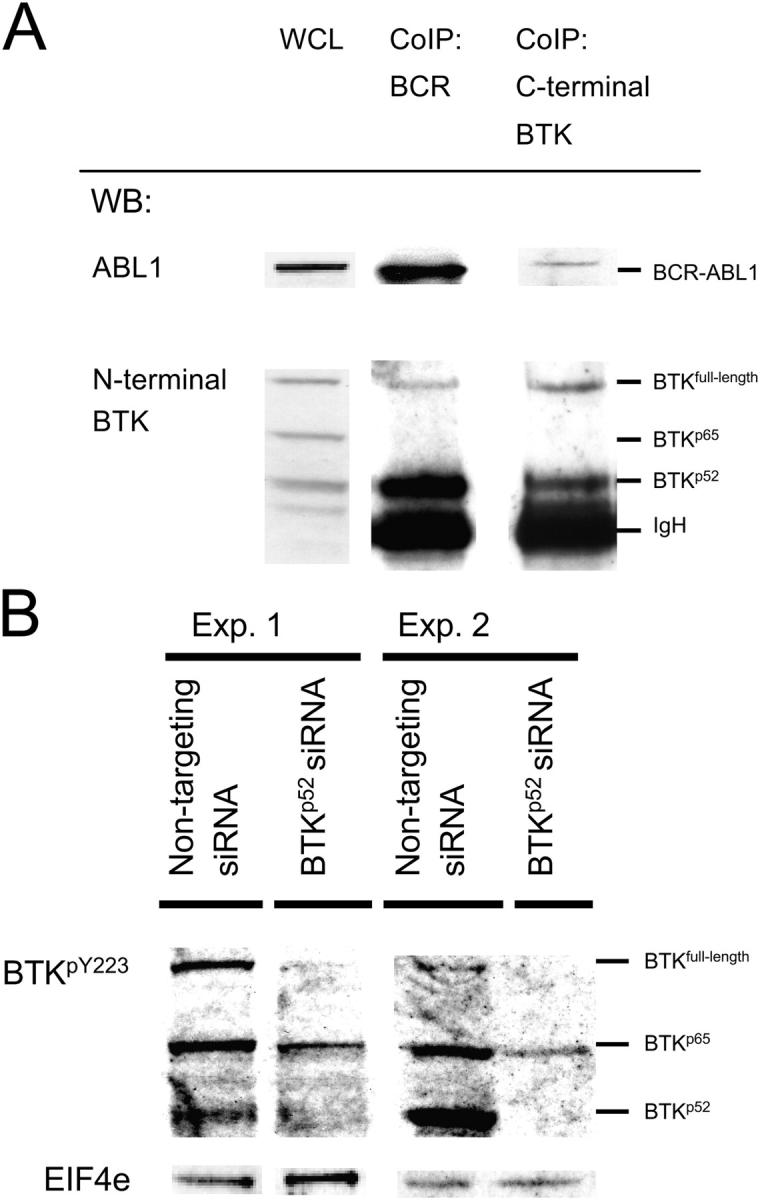
Pre-B cells undergo apoptosis unless they are rescued by pre-B cell receptor-dependent survival signals. We previously showed that the BCR-ABL1 kinase that is expressed in pre-B lymphoblastic leukemia bypasses selection for pre-B cell receptor-dependent survival signals. Investigating possible interference of BCR-ABL1 with pre-B cell receptor signaling, we found that neither SYK nor SLP65 can be phosphorylated in response to pre-B cell receptor engagement. Instead, Bruton's tyrosine kinase (BTK) is constitutively phosphorylated by BCR-ABL1. Activated BTK is essential for survival signals that otherwise would arise from the pre-B cell receptor, including activation of PLCgamma1, autonomous Ca2+ signaling, STAT5-phosphorylation, and up-regulation of BCLX(L). Inhibition of BTK activity specifically induces apoptosis in BCR-ABL1+ leukemia cells to a similar extent as inhibition of BCR-ABL1 kinase activity itself. However, BCR-ABL1 cannot directly bind to full-length BTK. Instead, BCR-ABL1 induces the expression of a truncated splice variant of BTK that acts as a linker between the two kinases. As opposed to full-length BTK, truncated BTK lacks kinase activity yet can bind to BCR-ABL1 through its SRC-homology domain 3. Acting as a linker, truncated BTK enables BCR-ABL1-dependent activation of full-length BTK, which initiates downstream survival signals and mimics a constitutively active pre-B cell receptor.
Figures







References
-
- Look, A.T. 1997. Oncogenic transcription factors in the human acute leukemias. Science. 278:1059–1064. - PubMed
-
- Huettner, C.S., P. Zhang, R.A. Van Etten, and D.G. Tenen. 2000. Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat. Genet. 24:57–60. - PubMed
-
- Rajewsky, K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758. - PubMed
-
- Kato, I., T. Takai, and A. Kudo. 2002. The pre-B cell receptor signaling for apoptosis is negatively regulated by Fc gamma RIIB. J. Immunol. 168:534–629. - PubMed
-
- Küppers, R., U. Klein, M.L. Hansmann, and K. Rajewsky. 1999. Cellular origin of human B-cell lymphomas. N. Engl. J. Med. 341:1520–1529. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
