

PAGE: parametric analysis of gene set enrichment
- PMID: 15941488
- PMCID: PMC1183189
- DOI: 10.1186/1471-2105-6-144
PAGE: parametric analysis of gene set enrichment
Abstract
Background: Gene set enrichment analysis (GSEA) is a microarray data analysis method that uses predefined gene sets and ranks of genes to identify significant biological changes in microarray data sets. GSEA is especially useful when gene expression changes in a given microarray data set is minimal or moderate.
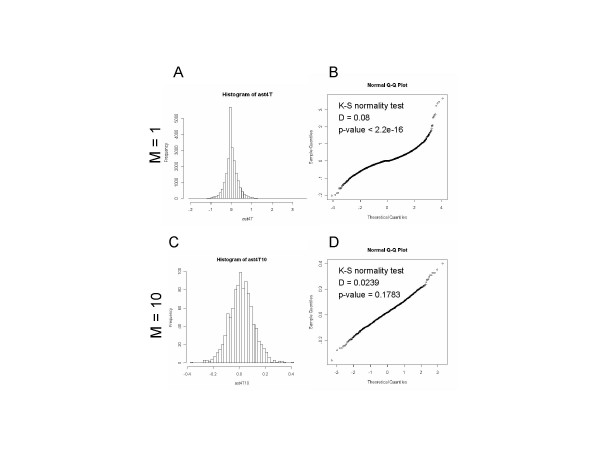
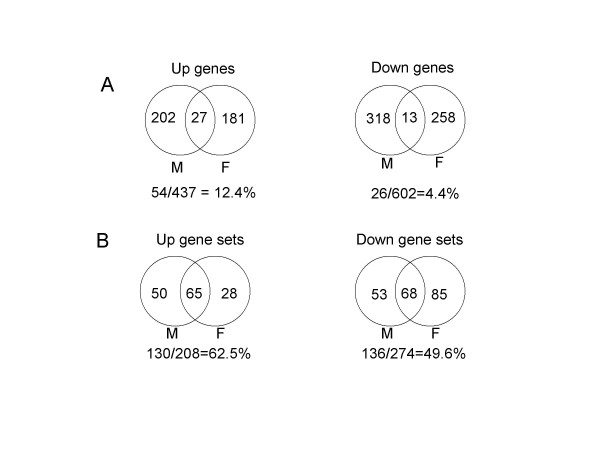
Results: We developed a modified gene set enrichment analysis method based on a parametric statistical analysis model. Compared with GSEA, the parametric analysis of gene set enrichment (PAGE) detected a larger number of significantly altered gene sets and their p-values were lower than the corresponding p-values calculated by GSEA. Because PAGE uses normal distribution for statistical inference, it requires less computation than GSEA, which needs repeated computation of the permutated data set. PAGE was able to detect significantly changed gene sets from microarray data irrespective of different Affymetrix probe level analysis methods or different microarray platforms. Comparison of two aged muscle microarray data sets at gene set level using PAGE revealed common biological themes better than comparison at individual gene level.
Conclusion: PAGE was statistically more sensitive and required much less computational effort than GSEA, it could identify significantly changed biological themes from microarray data irrespective of analysis methods or microarray platforms, and it was useful in comparison of multiple microarray data sets. We offer PAGE as a useful microarray analysis method.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
