

Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion
- PMID: 15958495
- PMCID: PMC1182326
- DOI: 10.1091/mbc.e04-12-1063
Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister-chromatid cohesion
Abstract
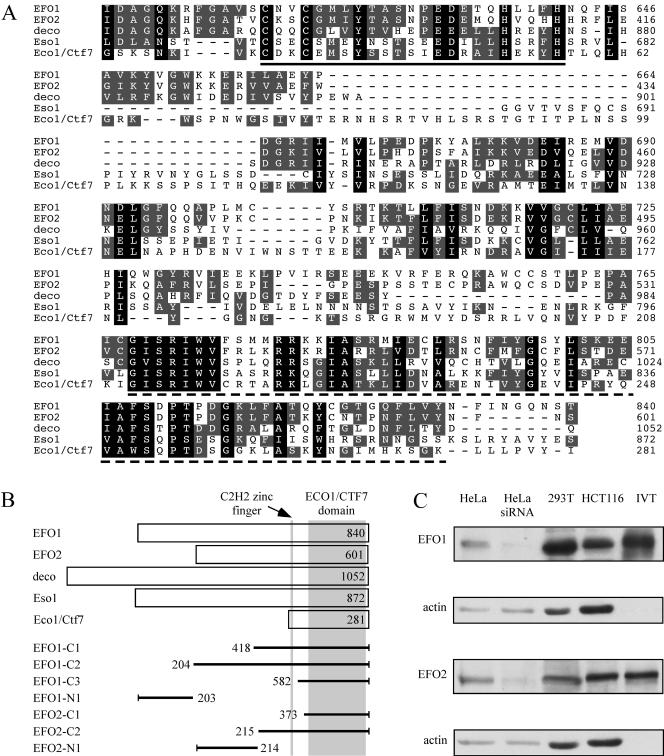
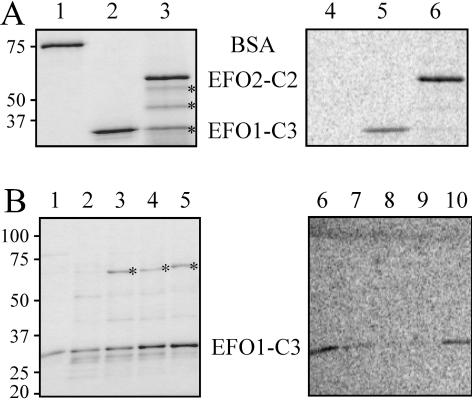
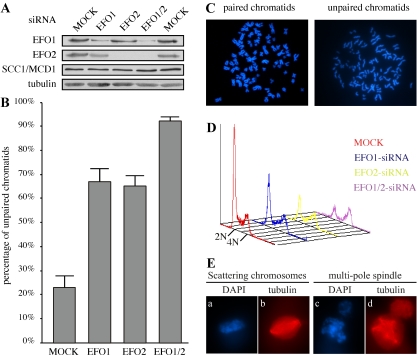
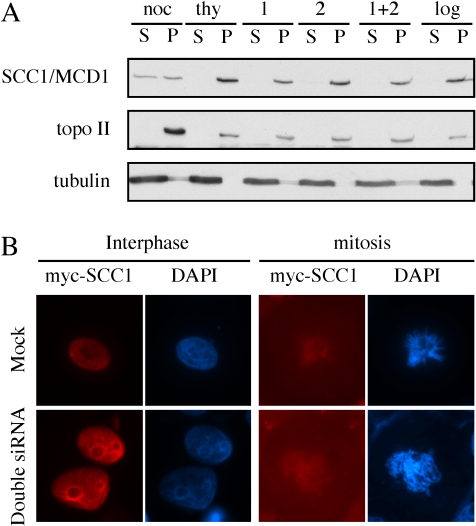
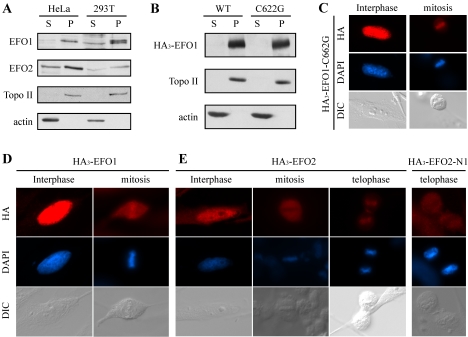
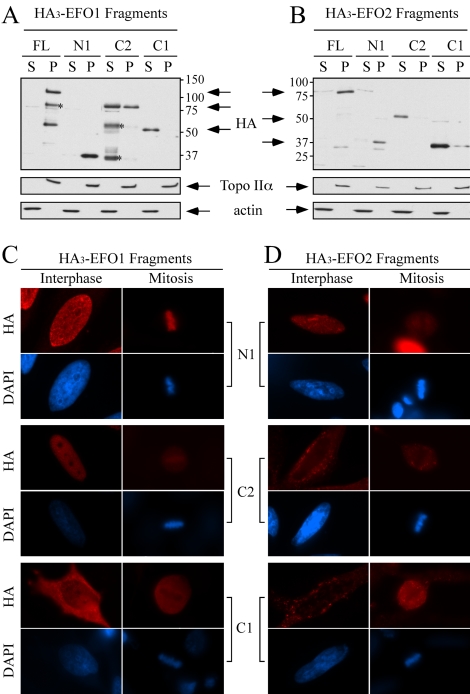
Genetic studies in yeast and Drosophila have uncovered a conserved acetyltransferase involved in sister-chromatid cohesion. Here, we described the two human orthologues, previously named EFO1/ESCO1 and EFO2/ESCO2. Similar to their yeast (Eco1/Ctf7 and Eso1) and fly (deco) counterparts, both proteins feature a conserved C-terminal domain consisting of a H2C2 zinc finger motif and an acetyltransferase domain that is able to catalyze autoacetylation reaction in vitro. However, no similarity can be detected outside of the conserved domain. RNA interference depletion experiment revealed that EFO1/ESCO1 and EFO2/ESCO2 were not redundant and that both were required for proper sister-chromatid cohesion. The difference between EFO1 and EFO2 also is reflected in their cell cycle regulation. In mitosis, EFO1 is phosphorylated, whereas EFO2 is degraded. Furthermore, both proteins associate with chromosomes, and the chromosome binding depends on the diverse N-terminal domains. We propose that EFO1 and EFO2 are targeted to different chromosome structures to help establish or maintain sister-chromatid cohesion.
Figures

Similar articles
-
Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila.Curr Biol. 2003 Dec 2;13(23):2025-36. doi: 10.1016/j.cub.2003.11.018. Curr Biol. 2003. PMID: 14653991
-
PCNA controls establishment of sister chromatid cohesion during S phase.Mol Cell. 2006 Sep 1;23(5):723-32. doi: 10.1016/j.molcel.2006.07.007. Epub 2006 Aug 24. Mol Cell. 2006. PMID: 16934511
-
The acetyltransferase Eco1 elicits cohesin dimerization during S phase.J Biol Chem. 2020 May 29;295(22):7554-7565. doi: 10.1074/jbc.RA120.013102. Epub 2020 Apr 20. J Biol Chem. 2020. PMID: 32312753 Free PMC article.
-
Fork it over: the cohesion establishment factor Ctf7p and DNA replication.J Cell Sci. 2007 Aug 1;120(Pt 15):2471-7. doi: 10.1242/jcs.011999. J Cell Sci. 2007. PMID: 17646671 Review.
-
The sister bonding of duplicated chromosomes.Semin Cell Dev Biol. 2011 Aug;22(6):566-71. doi: 10.1016/j.semcdb.2011.03.013. Epub 2011 Apr 7. Semin Cell Dev Biol. 2011. PMID: 21497666 Free PMC article. Review.
Cited by
-
Sequencing of candidate chromosome instability genes in endometrial cancers reveals somatic mutations in ESCO1, CHTF18, and MRE11A.PLoS One. 2013 Jun 3;8(6):e63313. doi: 10.1371/journal.pone.0063313. Print 2014. PLoS One. 2013. PMID: 23755103 Free PMC article.
-
Radiation-induced cellular senescence results from a slippage of long-term G2 arrested cells into G1 phase.Cell Cycle. 2013 May 1;12(9):1424-32. doi: 10.4161/cc.24528. Epub 2013 Apr 9. Cell Cycle. 2013. PMID: 23574719 Free PMC article.
-
Localization and regulation of murine Esco2 during male and female meiosis.Biol Reprod. 2012 Sep 13;87(3):61. doi: 10.1095/biolreprod.112.099978. Print 2012 Sep. Biol Reprod. 2012. PMID: 22699483 Free PMC article.
-
Symmetric control of sister chromatid cohesion establishment.Nucleic Acids Res. 2023 Jun 9;51(10):4760-4773. doi: 10.1093/nar/gkad146. Nucleic Acids Res. 2023. PMID: 36912084 Free PMC article.
-
The correlation of ESCO1 expression with a prognosis of prostate cancer and anti-tumor effect of ESCO1 silencing.Transl Cancer Res. 2019 Jun;8(3):950-961. doi: 10.21037/tcr.2019.05.34. Transl Cancer Res. 2019. PMID: 35116834 Free PMC article.
References
-
- Brands, A., and Skibbens, R. V. (2005). Ctf7p/Eco1p exhibits acetyltransferase activity–but does it matter? Curr. Biol. 15, R50-R51. - PubMed
-
- Ciosk, R., Shirayama, M., Shevchenko, A., Tanaka, T., Toth, A., and Nasmyth, K. (2000). Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol. Cell 5, 243-254. - PubMed
-
- Edwards, S., Li, C. M., Levy, D. L., Brown, J., Snow, P. M., and Campbell, J. L. (2003). Saccharomyces cerevisiae DNA polymerase epsilon and polymerase sigma interact physically and functionally, suggesting a role for polymerase epsilon in sister chromatid cohesion. Mol. Cell. Biol. 23, 2733-2748. - PMC - PubMed
-
- Fang, G., Yu, H., and Kirschner, M. W. (1998). Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell 2, 163-171. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
