

Single nucleotide extension technology for quantitative site-specific evaluation of metC/C in GC-rich regions
- PMID: 15958788
- PMCID: PMC1150895
- DOI: 10.1093/nar/gni094
Single nucleotide extension technology for quantitative site-specific evaluation of metC/C in GC-rich regions
Abstract
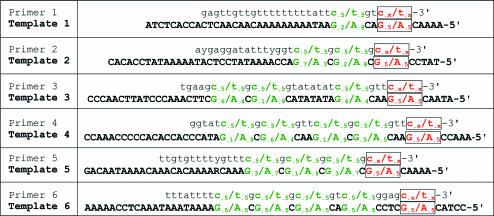
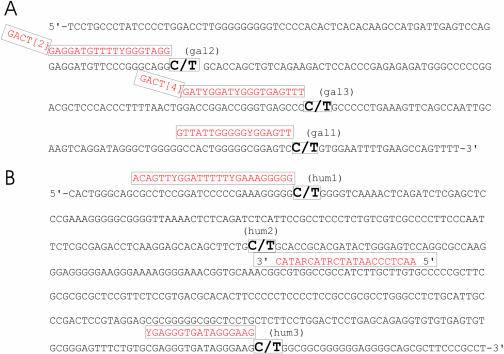
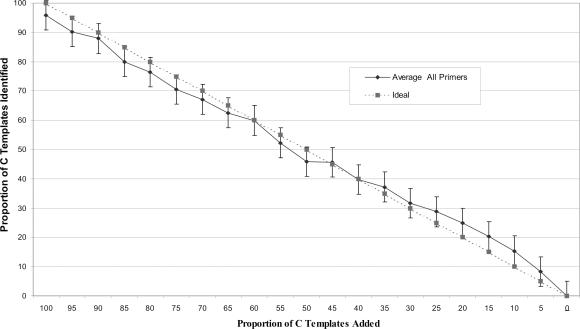
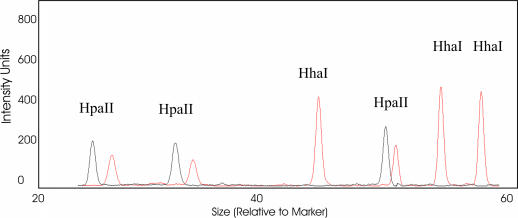
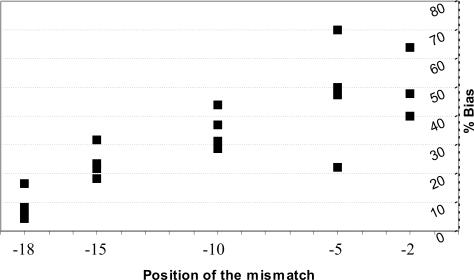
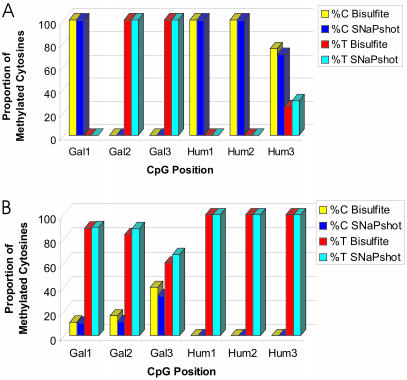
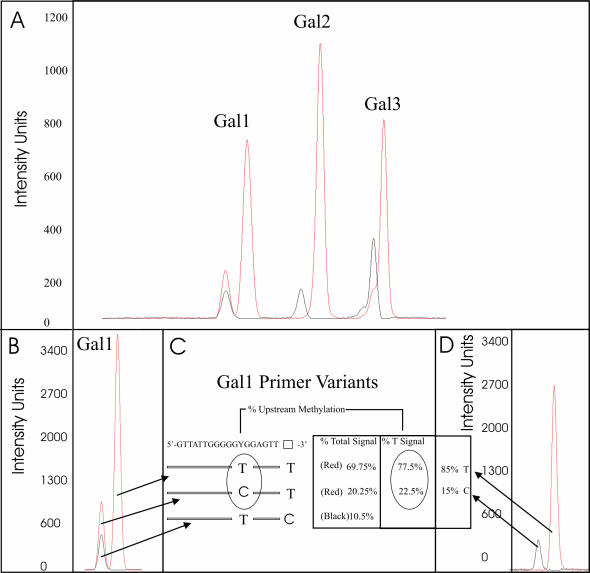
The development and use of high throughput technologies for detailed mapping of methylated cytosines (metC) is becoming of increasing importance for the expanding field of epigenetics. The single nucleotide primer extension reaction used for genotyping of single nucleotide polymorphisms has been recently adapted to interrogate the bisulfite modification induced 'quantitative' C/T polymorphism that corresponds to metC/C in the native DNA. In this study, we explored the opportunity to investigate C/T (and G/A) ratios using the Applied Biosystems (ABI) SNaPshot technology. The main effort of this study was dedicated to addressing the complexities in the analysis of DNA methylation in GC-rich regions where interrogation of the target cytosine can be confounded by variable degrees of methylation in other cytosines (resulting in variable C/T or G/A ratios after treatment with bisulfite) in the annealing site of the interrogating primer. In our studies, the mismatches of the SNaPshot primer with the target DNA sequence resulted in a biasing effect of up to 70% while these effects decreased as the location of the polymorphic site moved upstream of the target cytosine. We demonstrated that the biasing effect can be corrected with the SNaPshot primers containing degenerative C/T and G/A nucleotides. A series of experiments using various permutations of quantitative C/T and G/A polymorphisms at various positions of the target DNA sequence demonstrated that SNaPshot is able to accurately report cytosine methylation levels with <5% average SD from the true values. Given the relative simplicity of the method and the possibility to multiplex C/T and G/A interrogations, the SNaPshot approach may become a useful tool for large-scale mapping of metC.
Figures



Similar articles
-
Methylation SNaPshot: a method for the quantification of site-specific DNA methylation levels.Methods Mol Biol. 2009;507:241-55. doi: 10.1007/978-1-59745-522-0_18. Methods Mol Biol. 2009. PMID: 18987819
-
Methylation-sensitive single-nucleotide primer extension (Ms-SNuPE) for quantitative measurement of DNA methylation.Nat Protoc. 2007;2(8):1931-6. doi: 10.1038/nprot.2007.271. Nat Protoc. 2007. PMID: 17703204
-
Capillary electrophoretic analysis of methylation status in CpG-rich regions by single-base extension of primers modified with N6-methoxy-2,6-diaminopurine.Anal Biochem. 2008 Sep 1;380(1):13-20. doi: 10.1016/j.ab.2008.05.017. Epub 2008 May 20. Anal Biochem. 2008. PMID: 18539128
-
Genome-wide analysis of DNA methylation patterns.Development. 2007 Nov;134(22):3959-65. doi: 10.1242/dev.001131. Epub 2007 Oct 10. Development. 2007. PMID: 17928417 Review.
-
BiSearch: ePCR tool for native or bisulfite-treated genomic template.Methods Mol Biol. 2007;402:385-402. doi: 10.1007/978-1-59745-528-2_20. Methods Mol Biol. 2007. PMID: 17951807 Review.
Cited by
-
Epigenetic aging in patients diagnosed with coronary artery disease: results of the LipidCardio study.Clin Epigenetics. 2023 Jan 31;15(1):16. doi: 10.1186/s13148-023-01434-8. Clin Epigenetics. 2023. PMID: 36721243 Free PMC article.
-
Allelic skewing of DNA methylation is widespread across the genome.Am J Hum Genet. 2010 Feb 12;86(2):196-212. doi: 10.1016/j.ajhg.2010.01.014. Am J Hum Genet. 2010. PMID: 20159110 Free PMC article.
-
A simple algorithm for quantifying DNA methylation levels on multiple independent CpG sites in bisulfite genomic sequencing electropherograms.Nucleic Acids Res. 2008 Jun;36(11):e64. doi: 10.1093/nar/gkn210. Epub 2008 May 14. Nucleic Acids Res. 2008. PMID: 18480118 Free PMC article.
-
Simplified Assay for Epigenetic Age Estimation in Whole Blood of Adults.Front Genet. 2016 Jul 14;7:126. doi: 10.3389/fgene.2016.00126. eCollection 2016. Front Genet. 2016. PMID: 27471517 Free PMC article.
-
Vitamin D supplementation is associated with slower epigenetic aging.Geroscience. 2022 Jun;44(3):1847-1859. doi: 10.1007/s11357-022-00581-9. Epub 2022 May 13. Geroscience. 2022. PMID: 35562603 Free PMC article.
References
-
- Dahl C., Guldberg P. DNA methylation analysis techniques. Biogerontology. 2003;4:233–250. - PubMed
-
- Petronis A., Gottesman II, Kan P., Kennedy J.L., Basile V.S., Paterson A.D., Popendikyte V. Monozygotic twins exhibit numerous epigenetic differences: clues to twin discordance? Schizophr. Bull. 2003;29:169–178. - PubMed
-
- Gruenbaum Y., Stein R., Cedar H., Razin A. Methylation of CpG sequences in eukaryotic DNA. FEBS Lett. 1981;124:67–71. - PubMed
-
- Clark S.J., Harrison J., Frommer M. CpNpG methylation in mammalian cells. Nature Genet. 1995;10:20–27. - PubMed
-
- Norton N., Williams N.M., Williams H.J., Spurlock G., Kirov G., Morris D.W., Hoogendoorn B., Owen M.J., O'Donovan M.C. Universal, robust, highly quantitative SNP allele frequency measurement in DNA pools. Hum. Genet. 2002;110:471–478. - PubMed
