

Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1
- PMID: 15964798
- PMCID: PMC1156987
- DOI: 10.1128/MCB.25.13.5404-5416.2005
Transcription factor NF-kappaB differentially regulates death receptor 5 expression involving histone deacetylase 1
Abstract
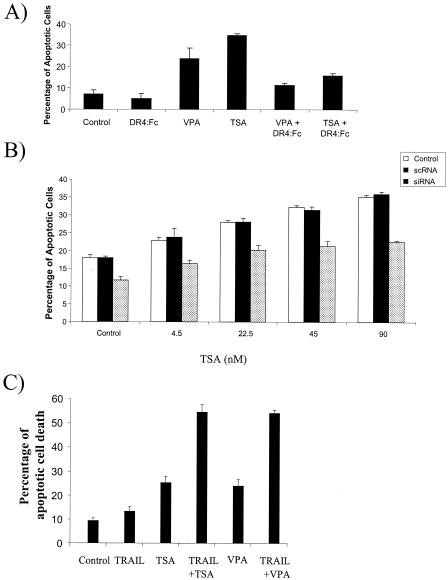
The transcription factor nuclear factor kappaB (NF-kappaB) regulates the expression of both anti-apoptotic and proapoptotic genes. Death receptor 5 (DR5, TRAIL-R2) is a proapoptotic protein considered to be a potential target for cancer therapy, and its expression is mediated by NF-kappaB. The mechanism of NF-kappaB-induced DR5 expression is, however, unknown. Herein, we determined that etoposide-induced DR5 expression requires the first intronic region of the DR5 gene. Mutation of a putative NF-kappaB binding site in this intron eliminates DR5 promoter activity, as do mutations in the p53 binding site in this region. Reduction in p53 expression also blocks p65 binding to the intronic region of the DR5 gene, indicating cooperation between p53 and p65 in DR5 expression. In contrast, the anti-apoptotic stimulus, epidermal growth factor (EGF), fails to increase DR5 expression but effectively activates NF-kappaB and induces p65 binding to the DR5 gene. EGF, however, induces the association of histone deacetylase 1 (HDAC1) with the DR5 gene, whereas etoposide treatment fails to induce this association. Indeed, HDAC inhibitors activate NF-kappaB and p53 and upregulate DR5 expression. Blockage of DR5 activation decreased HDAC inhibitor-induced apoptosis, and a combination of HDAC inhibitors and TRAIL increased apoptosis. This provides a mechanism for regulating NF-kappaB-mediated DR5 expression and could explain the differential roles NF-kappaB plays in regulating apoptosis.
Figures








References
-
- Aron, J. L., M. R. Parthun, G. Marcucci, S. Kitada, A. P. Mone, M. E. Davis, T. Shen, T. Murphy, J. Wickham, C. Kanakry, D. M. Lucas, J. C. Reed, M. R. Grever, and J. C. Byrd. 2003. Depsipeptide (FR901228) induces histone acetylation and inhibition of histone deacetylase in chronic lymphocytic leukemia cells concurrent with activation of caspase 8-mediated apoptosis and down-regulation of c-FLIP protein. Blood 102:652-658. - PubMed
-
- Ashkenazi, A., and V. M. Dixit. 1998. Death receptors: signaling and modulation. Science 281:1305-1308. - PubMed
-
- Baldwin, A. S., Jr. 1996. The NF-κB and IκB proteins: new discoveries and insights. Annu. Rev. Immunol. 14:649-683. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous