

Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity
- PMID: 15965030
- PMCID: PMC1172042
- DOI: 10.1101/gr.3804205
Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity
Abstract
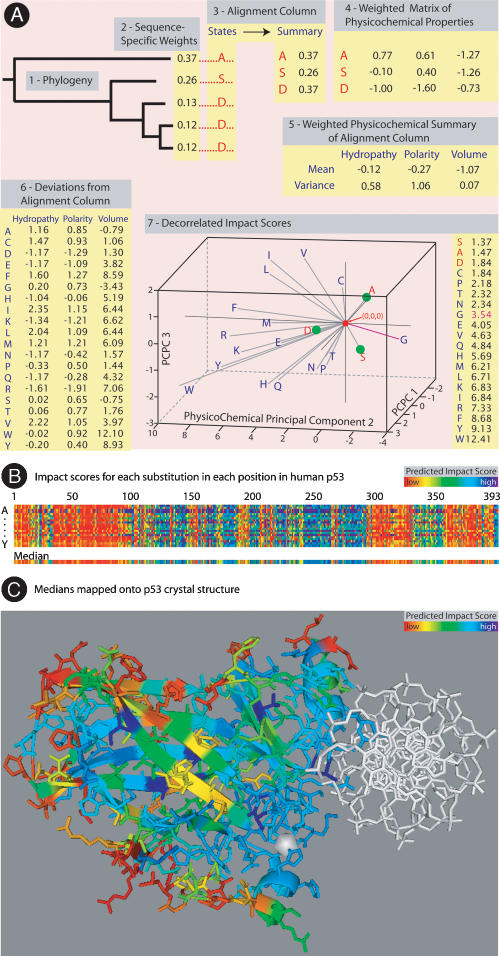
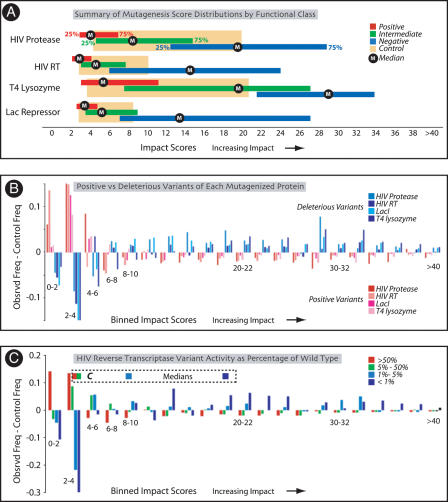
We find that the degree of impairment of protein function by missense variants is predictable by comparative sequence analysis alone. The applicable range of impairment is not confined to binary predictions that distinguish normal from deleterious variants, but extends continuously from mild to severe effects. The accuracy of predictions is strongly dependent on sequence variation and is highest when diverse orthologs are available. High predictive accuracy is achieved by quantification of the physicochemical characteristics in each position of the protein, based on observed evolutionary variation. The strong relationship between physicochemical characteristics of a missense variant and impairment of protein function extends to human disease. By using four diverse proteins for which sufficient comparative sequence data are available, we show that grades of disease, or likelihood of developing cancer, correlate strongly with physicochemical constraint violation by causative amino acid variants.
Figures



References
-
- Altschul, S.F., Carroll, R.J., and Lipman, D.J. 1989. Weights for data related by a tree. J. Mol. Biol. 207: 647-653. - PubMed
-
- Botstein, D. and Risch, N. 2003. Discovering genotypes underlying human phenotypes: Past successes for mendelian disease, future approaches for complex disease. Nat. Genet. 33(Suppl): 228-237. - PubMed
-
- Cai, Z., Tsung, E.F., Marinescu, V.D., Ramoni, M.F., Riva, A., and Kohane, I.S. 2004. Bayesian approach to discovering pathogenic SNPs in conserved protein domains. Hum. Mutat. 24: 178-184. - PubMed
-
- Cho, Y., Gorina, S., Jeffrey, P.D., and Pavletich, N.P. 1994. Crystal structure of a p53 tumor suppressor–DNA complex: Understanding tumorigenic mutations. Science 265: 346-355. - PubMed
-
- Coffin, J.M. 1995. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 267: 483-488. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases