

Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid
- PMID: 15967035
- PMCID: PMC1190207
- DOI: 10.1186/1742-2094-2-15
Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid
Abstract
Background: Interleukin 1 (IL-1) is a key mediator of immune responses in health and disease. Although classically the function of IL-1 has been studied in the systemic immune system, research in the past decade has revealed analogous roles in the CNS where the cytokine can contribute to the neuroinflammation and neuropathology seen in a number of neurodegenerative diseases. In Alzheimer's disease (AD), for example, pre-clinical and clinical studies have implicated IL-1 in the progression of a pathologic, glia-mediated pro-inflammatory state in the CNS. The glia-driven neuroinflammation can lead to neuronal damage, which, in turn, stimulates further glia activation, potentially propagating a detrimental cycle that contributes to progression of pathology. A prediction of this neuroinflammation hypothesis is that increased IL-1 signaling in vivo would correlate with increased severity of AD-relevant neuroinflammation and neuronal damage.
Methods: To test the hypothesis that increased IL-1 signaling predisposes animals to beta-amyloid (Abeta)-induced damage, we used IL-1 receptor antagonist Knock-Out (IL1raKO) and wild-type (WT) littermate mice in a model that involves intracerebroventricular infusion of human oligomeric Abeta1-42. This model mimics many features of AD, including robust neuroinflammation, Abeta plaques, synaptic damage and neuronal loss in the hippocampus. IL1raKO and WT mice were infused with Abeta for 28 days, sacrificed at 42 days, and hippocampal endpoints analyzed.
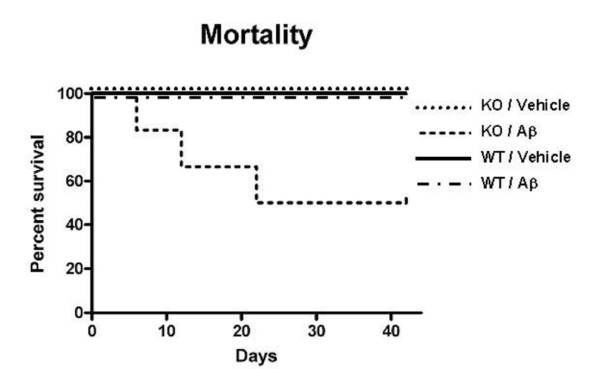
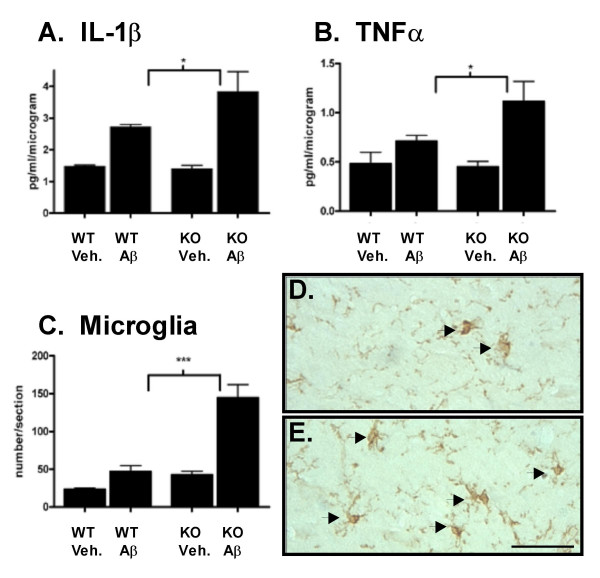
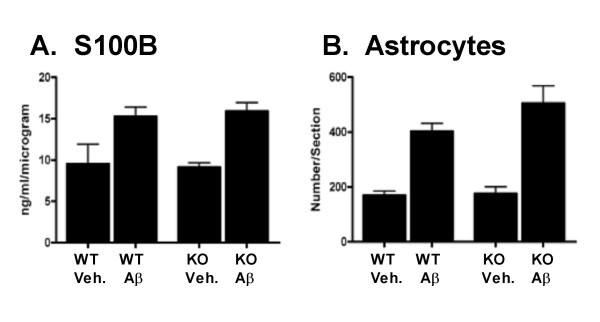
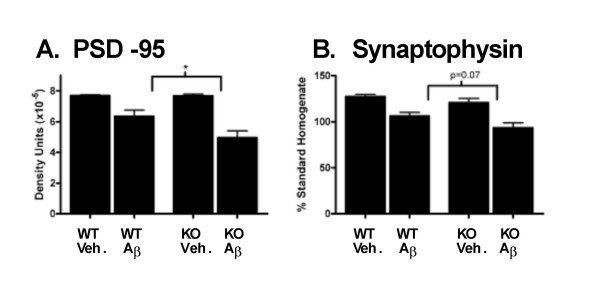
Results: IL1raKO mice showed increased vulnerability to Abeta-induced neuropathology relative to their WT counterparts. Specifically, IL1raKO mice exhibited increased mortality, enhanced microglial activation and neuroinflammation, and more pronounced loss of synaptic markers. Interestingly, Abeta-induced astrocyte responses were not significantly different between WT and IL1raKO mice, suggesting that enhanced IL-1 signaling predominately affects microglia.
Conclusion: Our data are consistent with the neuroinflammation hypothesis whereby increased IL-1 signaling in AD enhances glia activation and leads to an augmented neuroinflammatory process that increases the severity of neuropathologic sequelae.
Figures
Similar articles
-
Enhanced susceptibility of S-100B transgenic mice to neuroinflammation and neuronal dysfunction induced by intracerebroventricular infusion of human beta-amyloid.Glia. 2005 Aug 15;51(3):209-16. doi: 10.1002/glia.20194. Glia. 2005. PMID: 15810011
-
B₂ receptor blockage prevents Aβ-induced cognitive impairment by neuroinflammation inhibition.Behav Brain Res. 2015 Feb 1;278:482-91. doi: 10.1016/j.bbr.2014.10.040. Epub 2014 Nov 3. Behav Brain Res. 2015. PMID: 25446751
-
Cooperative therapeutic action of retinoic acid receptor and retinoid x receptor agonists in a mouse model of Alzheimer's disease.J Alzheimers Dis. 2014;42(2):587-605. doi: 10.3233/JAD-132720. J Alzheimers Dis. 2014. PMID: 24916544
-
The contribution of neuroinflammation to amyloid toxicity in Alzheimer's disease.J Neurochem. 2016 Feb;136(3):457-74. doi: 10.1111/jnc.13411. Epub 2015 Nov 18. J Neurochem. 2016. PMID: 26509334 Review.
-
Physical activity and exercise attenuate neuroinflammation in neurological diseases.Brain Res Bull. 2016 Jul;125:19-29. doi: 10.1016/j.brainresbull.2016.03.012. Epub 2016 Mar 26. Brain Res Bull. 2016. PMID: 27021169 Review.
Cited by
-
Cytokines and Cytokine Receptors Involved in the Pathogenesis of Alzheimer's Disease.J Clin Cell Immunol. 2016 Aug;7(4):441. doi: 10.4172/2155-9899.1000441. Epub 2016 Aug 4. J Clin Cell Immunol. 2016. PMID: 27895978 Free PMC article.
-
Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer's Disease.Neurochem Res. 2020 Nov;45(11):2560-2572. doi: 10.1007/s11064-020-03121-z. Epub 2020 Sep 14. Neurochem Res. 2020. PMID: 32929691 Review.
-
Cotinine and 6-Hydroxy-L-Nicotine Reverses Memory Deficits and Reduces Oxidative Stress in Aβ25-35-Induced Rat Model of Alzheimer's Disease.Antioxidants (Basel). 2020 Aug 18;9(8):768. doi: 10.3390/antiox9080768. Antioxidants (Basel). 2020. PMID: 32824768 Free PMC article.
-
Neuroinflammatory Triangle Presenting Novel Pharmacological Targets for Ischemic Brain Injury.Front Immunol. 2021 Oct 7;12:748663. doi: 10.3389/fimmu.2021.748663. eCollection 2021. Front Immunol. 2021. PMID: 34691061 Free PMC article. Review.
-
The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective.J Neuroinflammation. 2008 Feb 26;5:7. doi: 10.1186/1742-2094-5-7. J Neuroinflammation. 2008. PMID: 18302763 Free PMC article. Review.
References
-
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/S0197-4580(00)00124-X. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
