

A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma
- PMID: 15968592
- PMCID: PMC1224527
- DOI: 10.1086/432556
A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma
Abstract
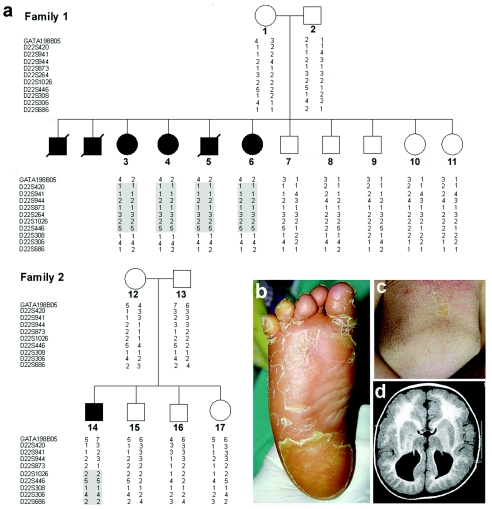
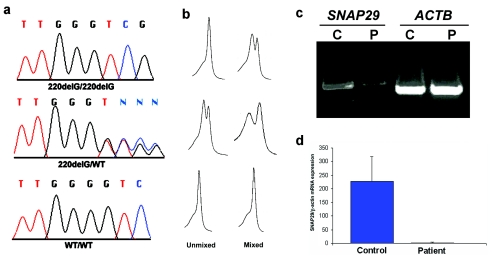
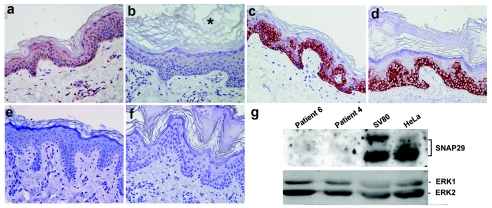
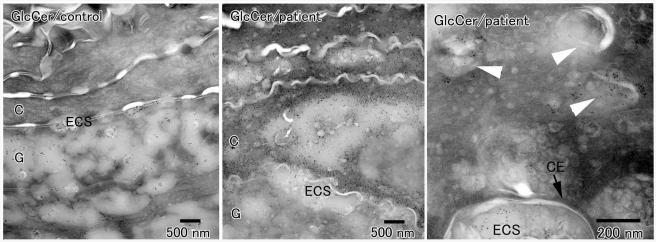
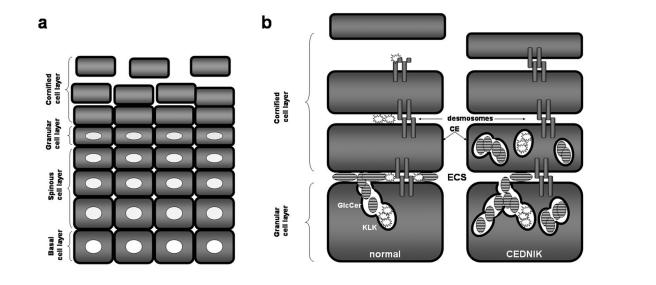
Neurocutaneous syndromes represent a vast, largely heterogeneous group of disorders characterized by neurological and dermatological manifestations, reflecting the common embryonic origin of epidermal and neural tissues. In the present report, we describe a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and keratoderma (CEDNIK syndrome). Using homozygosity mapping in two large families, we localized the disease gene to 22q11.2 and identified, in all patients, a 1-bp deletion in SNAP29, which codes for a SNARE protein involved in vesicle fusion. SNAP29 expression was decreased in the skin of the patients, resulting in abnormal maturation of lamellar granules and, as a consequence, in mislocation of epidermal lipids and proteases. These data underscore the importance of vesicle trafficking regulatory mechanisms for proper neuroectodermal differentiation.
Figures

References
Web Resources
-
- DHPLC Melt program, http://insertion.stanford.edu/meltdoc.html
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
-
- Superlink, http://bioinfo.cs.technion.ac.il/pedtool/
References
-
- Bercovich D, Beaudet AL (2003) Denaturing high-performance liquid chromatography for the detection of mutations and polymorphisms in UBE3A. Genet Test 7:189–194 - PubMed
-
- Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116:153–166 - PubMed
-
- Canaani D, Naiman T, Teitz T, Berg P (1986) Immortalization of xeroderma pigmentosum cells by simian virus 40 DNA having a defective origin of DNA replication. Somat Cell Mol Genet 12:13–20 - PubMed
-
- Caubet C, Jonca N, Brattsand M, Guerrin M, Bernard D, Schmidt R, Egelrud T, Simon M, Serre G (2004) Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J Invest Dermatol 122:1235–1244 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
