

Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing
- PMID: 15972952
- PMCID: PMC1603435
- DOI: 10.1016/s0002-9440(10)62953-7
Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing
Abstract
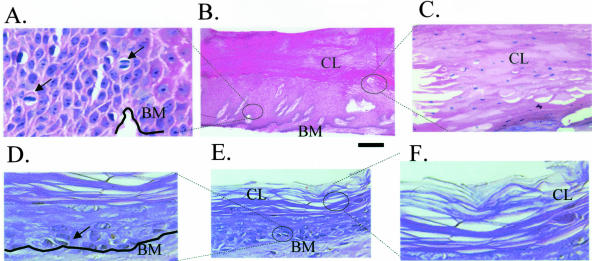
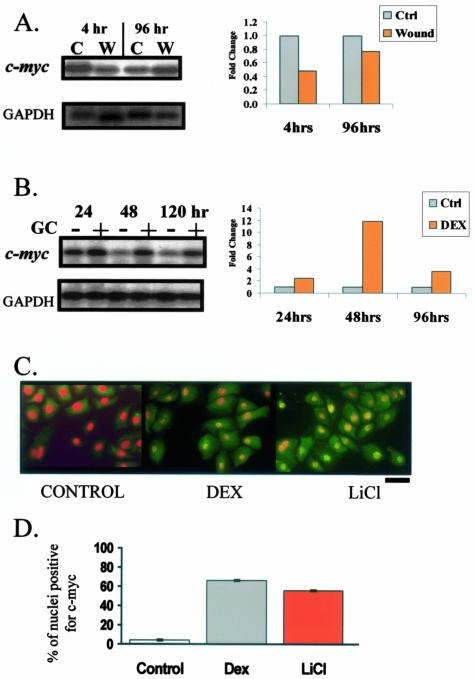
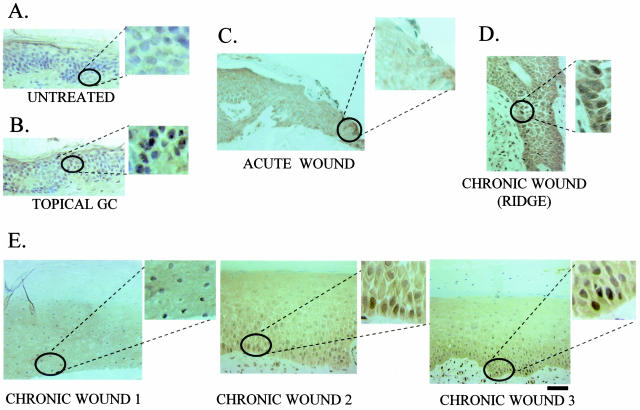
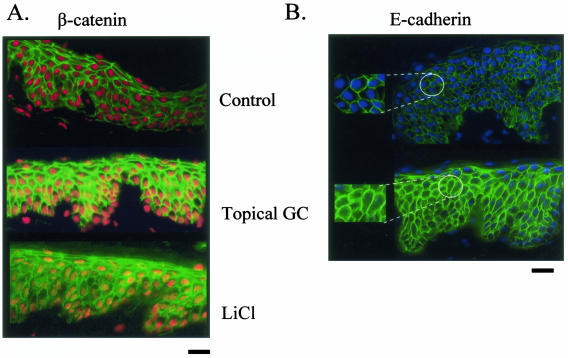
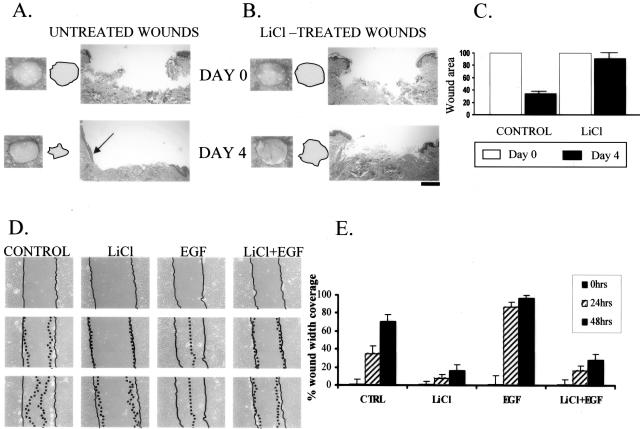
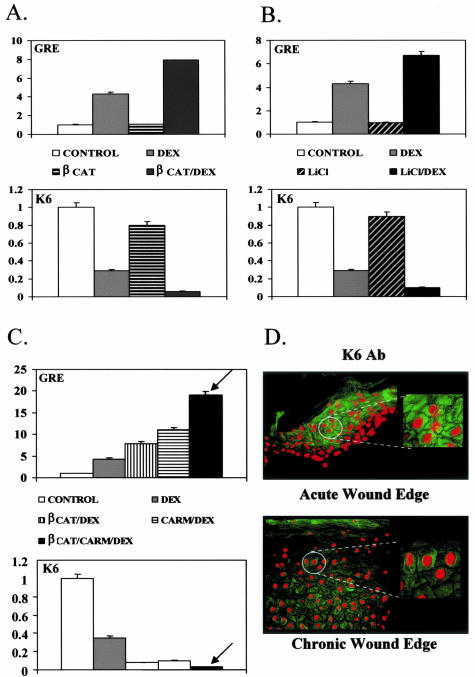
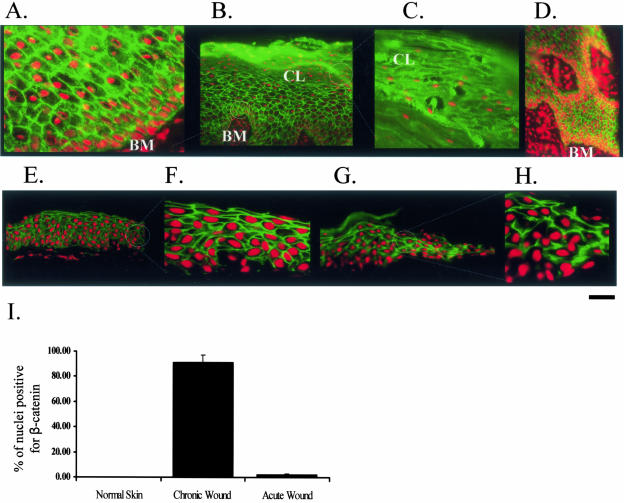
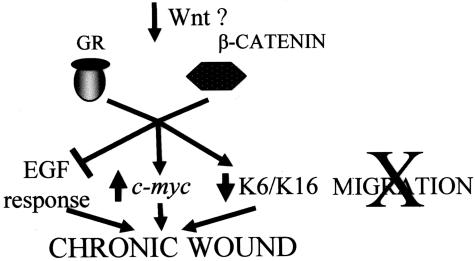
Lack of understanding of the molecular mechanisms and pathogenesis of impaired healing in chronic ulcers is a serious health issue that contributes to excessive limb amputations and mortality. Here we show that beta-catenin and its downstream targets in keratinocytes, c-myc, and keratins K6 and K16, play important roles in the development of chronic wounds. In contrast to normal epidermis, we observed a significant nuclear presence of beta-catenin and elevated c-myc expression at the nonhealing wound edge of chronic ulcers from 10 patients. In vitro studies indicated that stabilization of nuclear beta-catenin inhibited wound healing and keratinocyte migration by blocking epidermal growth factor response, inducing c-myc and repressing the K6/K16 keratins (cytoskeletal components important for migration). The molecular mechanism of K6/K16 repression involved beta-catenin and arginine methyltransferase (CARM-1) acting as co-repressors of glucocorticoid receptor monomers. We conclude that activation of the beta-catenin/c-myc pathway(s) contributes to impaired healing by inhibiting keratinocyte migration and altering their differentiation. The presence of activated beta-catenin and c-myc in the epidermis of chronic wounds may serve as a molecular marker of impaired healing and may provide future targets for therapeutic intervention.
Figures
References
-
- Tomic-Canic M, Magnus SA, Oscar MA. Epidermal repair and the chronic wound. Rovee DT, Maibach HI, editors. Boca Raton: CRC Press LLC,; The epidermis in wound healing. 2004:pp 25–57.
-
- Jeffcoate WJ, Harding KG. Diabetic foot ulcers. Lancet. 2003;361:1545–1551. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
