

Novel approach to mapping of resistance mutations in whole genomes by using restriction enzyme modulation of transformation efficiency
- PMID: 15980348
- PMCID: PMC1168657
- DOI: 10.1128/AAC.49.7.2767-2777.2005
Novel approach to mapping of resistance mutations in whole genomes by using restriction enzyme modulation of transformation efficiency
Abstract
Restriction enzyme modulation of transformation efficiencies (REMOTE) is a method that makes use of genome restriction maps and experimentally observed differences in transformation efficiencies of genomic DNA restriction digests to discover the location of mutations in genomes. The frequency with which digested genomic DNA from a resistant strain transforms a susceptible strain to resistance is primarily determined by the size of the fragment containing the resistance mutation and the distance of the mutation to the end of the fragment. The positions of restriction enzyme cleavage sites immediately flanking the resistance mutation define these parameters. The mapping procedure involves a process of elimination in which digests that transform with high frequency indicate that the restriction enzyme cleavage sites are relatively far away from the mutation, while digests that transform with low frequency indicate that the sites are close to the mutation. The transformation data are compared computationally to the genome restriction map to identify the regions that best fit the data. Transformations with PCR amplicons encompassing candidate regions identify the resistance locus and enable identification of the mutation. REMOTE was developed using Haemophilus influenzae strains with mutations in gyrA, gyrB, and rpsE that confer resistance to ciprofloxacin, novobiocin, and spectinomycin, respectively. We applied REMOTE to identify mutations that confer resistance to two novel antibacterial compounds. The resistance mutations were found in genes that can decrease the intracellular concentration of compounds: acrB, which encodes a subunit of the AcrAB-TolC efflux pump; and fadL, which encodes a long-chain fatty acid transporter.
Figures

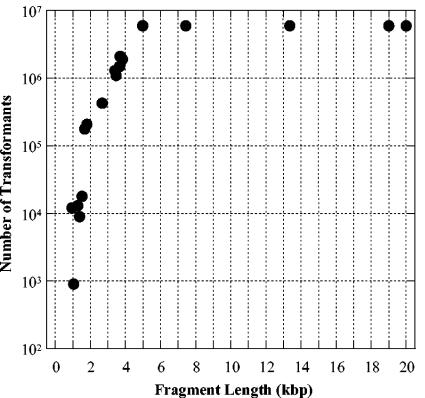
 ). The mutation is indicated by an inverse white circle (
). The mutation is indicated by an inverse white circle ( ). For a hypothetical genomic segment containing 10 genes, the data fit best to gene 3, which contains sites for both low-transforming digests (C and D) in close proximity and the sites for the high-transforming digests (A and B), which are further away. The other nine genes do not have clusters of the sites for both low-transforming digests that are also far from sites for the high-transforming digests.
). For a hypothetical genomic segment containing 10 genes, the data fit best to gene 3, which contains sites for both low-transforming digests (C and D) in close proximity and the sites for the high-transforming digests (A and B), which are further away. The other nine genes do not have clusters of the sites for both low-transforming digests that are also far from sites for the high-transforming digests.



References
-
- Abbanat, D., M. Macielag, and K. Bush. 2003. Novel antibacterial agents for the treatment of serious Gram-positive infections. Expert Opin. Investig. Drugs 12:379-399. - PubMed
-
- Andries, K., P. Verhasselt, J. Guillemont, H. W. Gohlmann, J. M. Neefs, H. Winkler, J. Van Gestel, P. Timmerman, M. Zhu, E. Lee, P. Williams, D. de Chaffoy, E. Huitric, S. Hoffner, E. Cambau, C. Truffot-Pernot, N. Lounis, and V. Jarlier. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223-227. - PubMed
-
- Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1991. Current protocols in molecular biology, p. 2.4.1-2.4.2. John Wiley & Sons, New York, N.Y.
-
- Babon, J. J., M. McKenzie, and R. G. Cotton. 2003. The use of resolvases T4 endonuclease VII and T7 endonuclease I in mutation detection. Mol. Biotechnol. 23:73-81. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
