

MAVL/StickWRLD for protein: visualizing protein sequence families to detect non-consensus features
- PMID: 15980480
- PMCID: PMC1160135
- DOI: 10.1093/nar/gki374
MAVL/StickWRLD for protein: visualizing protein sequence families to detect non-consensus features
Abstract
A fundamental problem with applying Consensus, Weight-Matrix or hidden Markov models as search tools for biosequences is that there is no way to know, from the model, if the modeled sequences display any dependencies between positional identities. In some instances, these dependencies are crucial in correctly accepting or rejecting other sequences as members of the family. MAVL (multiple alignment variation linker) and StickWRLD provide a web-based method to visually survey the model-training sequences to discover and characterize possible dependencies. Initially introduced for nucleic acid sequences, with MAVL/StickWRLD, it is easy to distinguish typical DNA or RNA structural dependencies in input families, identify mixed populations of distinct subfamilies, or discover novel dependencies that result from binding interactions or other selective pressures [W. Ray (2004) Nucleic Acids Res., 32, W59-W63]. Since the announcement of MAVL/StickWRLD for nucleic acids, one of the most requested new features has been the extension of this visualization method to support protein alignments. We are pleased to report that this extension has been successful, that the basic visualization has been augmented in several ways to enhance protein viewing, and that the results with protein alignments are even more dramatic than with NA alignments. MAVL/StickWRLD can be accessed at http://www.microbial-pathogenesis.org/stickwrld/.
Figures
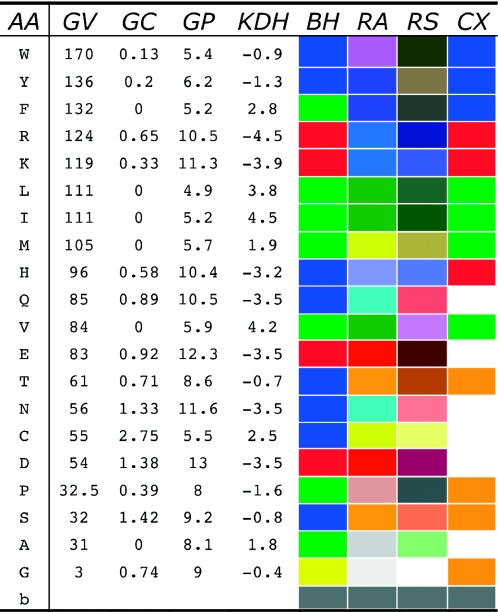


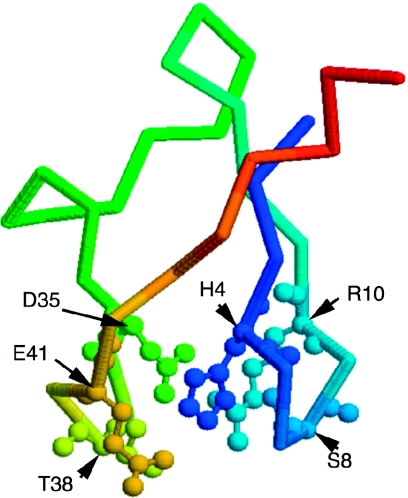
Similar articles
-
MAVL/StickWRLD: analyzing structural constraints using interpositional dependencies in biomolecular sequence alignments.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W133-6. doi: 10.1093/nar/gkl251. Nucleic Acids Res. 2006. PMID: 16844976 Free PMC article.
-
MAVL and StickWRLD: visually exploring relationships in nucleic acid sequence alignments.Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W59-63. doi: 10.1093/nar/gkh469. Nucleic Acids Res. 2004. PMID: 15215351 Free PMC article.
-
ViTO: tool for refinement of protein sequence-structure alignments.Bioinformatics. 2004 Dec 12;20(18):3694-6. doi: 10.1093/bioinformatics/bth429. Epub 2004 Jul 22. Bioinformatics. 2004. PMID: 15271783
-
MAGOS: multiple alignment and modelling server.Bioinformatics. 2006 Sep 1;22(17):2164-5. doi: 10.1093/bioinformatics/btl349. Epub 2006 Jul 4. Bioinformatics. 2006. PMID: 16820425
-
Discovering patterns and subfamilies in biosequences.Proc Int Conf Intell Syst Mol Biol. 1996;4:34-43. Proc Int Conf Intell Syst Mol Biol. 1996. PMID: 8877502 Review.
Cited by
-
MAVL/StickWRLD: analyzing structural constraints using interpositional dependencies in biomolecular sequence alignments.Nucleic Acids Res. 2006 Jul 1;34(Web Server issue):W133-6. doi: 10.1093/nar/gkl251. Nucleic Acids Res. 2006. PMID: 16844976 Free PMC article.
-
Optimization of Synthetic Proteins: Identification of Interpositional Dependencies Indicating Structurally and/or Functionally Linked Residues.J Vis Exp. 2015 Jul 14;(101):e52878. doi: 10.3791/52878. J Vis Exp. 2015. PMID: 26274377 Free PMC article.
-
FvatfA regulates growth, stress tolerance as well as mycotoxin and pigment productions in Fusarium verticillioides.Appl Microbiol Biotechnol. 2020 Sep;104(18):7879-7899. doi: 10.1007/s00253-020-10717-6. Epub 2020 Jul 27. Appl Microbiol Biotechnol. 2020. PMID: 32719911 Free PMC article.
-
Understanding the sequence requirements of protein families: insights from the BioVis 2013 contests.BMC Proc. 2014 Aug 28;8(Suppl 2 Proceedings of the 3rd Annual Symposium on Biologica):S1. doi: 10.1186/1753-6561-8-S2-S1. eCollection 2014. BMC Proc. 2014. PMID: 25237388 Free PMC article.
-
Addressing the unmet need for visualizing conditional random fields in biological data.BMC Bioinformatics. 2014 Jul 10;15:202. doi: 10.1186/1471-2105-15-202. BMC Bioinformatics. 2014. PMID: 25000815 Free PMC article.
References
-
- Bailey T.L., Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. - PubMed
-
- Durbin R., Eddy S., Krogh A., Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge: Cambridge University Press; 1998.
-
- Taylor W.R., Hatrick K. Compensating changes in protein multiple sequence alignments. Protein Eng. 1994;7:341–348. - PubMed
