

Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase "Asian" variant
- PMID: 15983043
- PMCID: PMC1262676
- DOI: 10.1074/jbc.M502345200
Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase "Asian" variant
Abstract
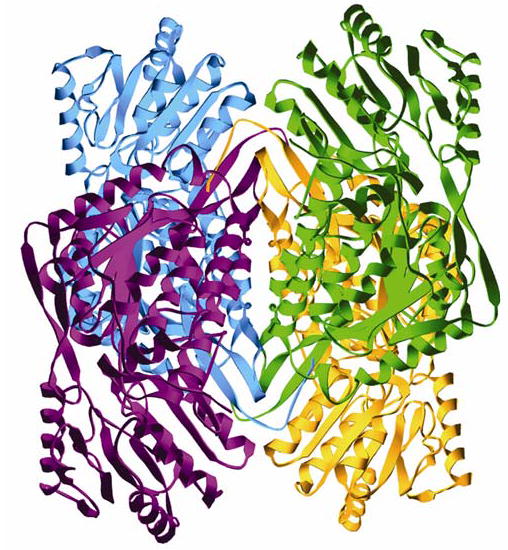
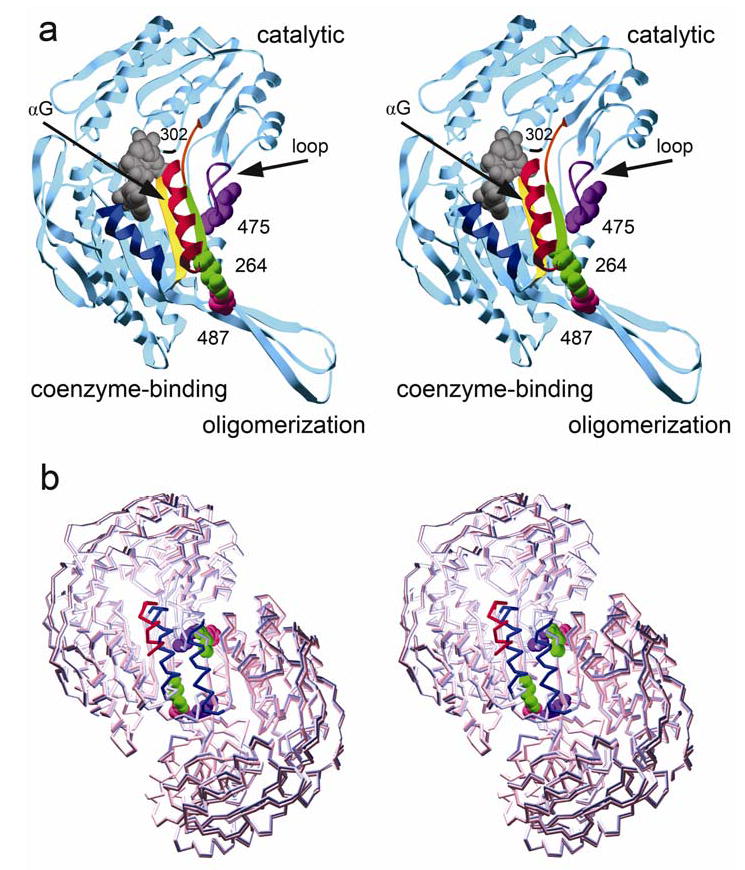
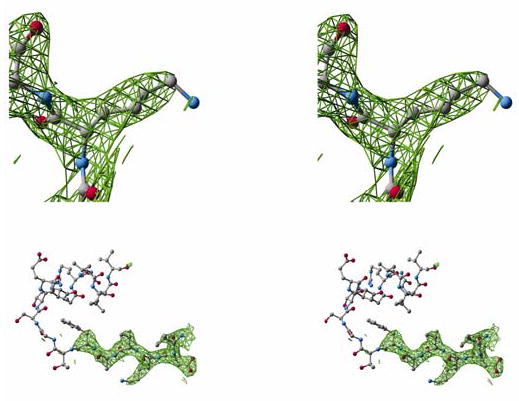
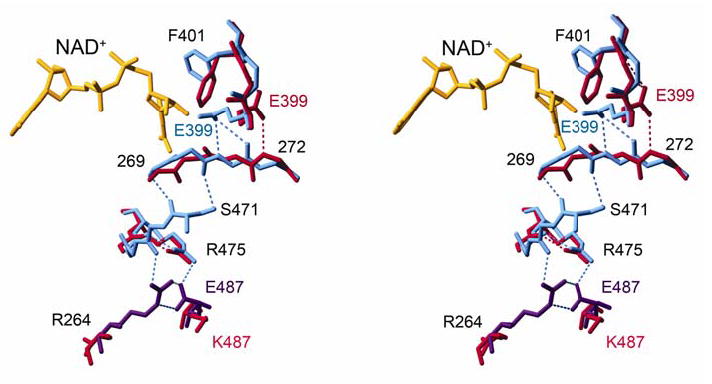
Mitochondrial aldehyde dehydrogenase (ALDH2) is the major enzyme that oxidizes ethanol-derived acetaldehyde. A nearly inactive form of the enzyme, ALDH2*2, is found in about 40% of the East Asian population. This variant enzyme is defined by a glutamate to lysine substitution at residue 487 located within the oligomerization domain. ALDH2*2 has an increased Km for its coenzyme, NAD+, and a decreased kcat, which lead to low activity in vivo. Here we report the 2.1 A crystal structure of ALDH2*2. The structure shows a large disordered region located at the dimer interface that includes much of the coenzyme binding cleft and a loop of residues that form the base of the active site. As a consequence of these structural changes, the variant enzyme exhibits rigid body rotations of its catalytic and coenzyme-binding domains relative to the oligomerization domain. These structural perturbations are the direct result of the inability of lysine 487 to form important stabilizing hydrogen bonds with arginines 264 and 475. Thus, the elevated Km for coenzyme exhibited by this variant probably reflects the energetic penalty for reestablishing this site for productive coenzyme binding, whereas the structural alterations near the active site are consistent with the lowered Vmax.
Figures
References
-
- Hurley, T. D., Edenberg, H. J., and Li, T. K. (2002) in Pharmacogenomics: The Search for Individualized Therapies (Licinio, J., and Wong, M., eds), pp. 417–441, Culinary and Hospitality Industry Publications Services
-
- Seitz HK, Matsuzaki S, Yokoyama A, Homann N, Vakevainen S, Wang XD. Alcoholism: Clinical & Experimental Research. 2001;25:137S–143S. - PubMed
-
- Peng GS, Wang MF, Chen CY, Luu SU, Chou HC, Li TK, Yin SJ. Pharmacogenetics. 1999;9:463–476. - PubMed
-
- Enomoto N, Takase S, Takada N, Takada A. Hepatology. 1991;13:1071–1075. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
