

Multiple sequence alignments of partially coding nucleic acid sequences
- PMID: 15985156
- PMCID: PMC1182351
- DOI: 10.1186/1471-2105-6-160
Multiple sequence alignments of partially coding nucleic acid sequences
Abstract
Background: High quality sequence alignments of RNA and DNA sequences are an important prerequisite for the comparative analysis of genomic sequence data. Nucleic acid sequences, however, exhibit a much larger sequence heterogeneity compared to their encoded protein sequences due to the redundancy of the genetic code. It is desirable, therefore, to make use of the amino acid sequence when aligning coding nucleic acid sequences. In many cases, however, only a part of the sequence of interest is translated. On the other hand, overlapping reading frames may encode multiple alternative proteins, possibly with intermittent non-coding parts. Examples are, in particular, RNA virus genomes.
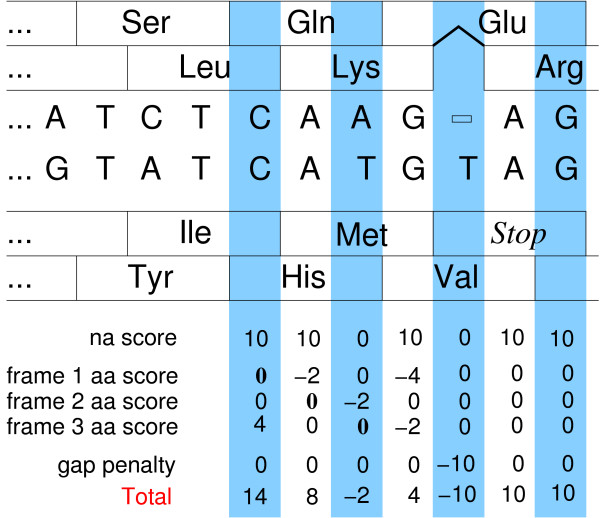
Results: The standard scoring scheme for nucleic acid alignments can be extended to incorporate simultaneously information on translation products in one or more reading frames. Here we present a multiple alignment tool, codaln, that implements a combined nucleic acid plus amino acid scoring model for pairwise and progressive multiple alignments that allows arbitrary weighting for almost all scoring parameters. Resource requirements of codaln are comparable with those of standard tools such as ClustalW.
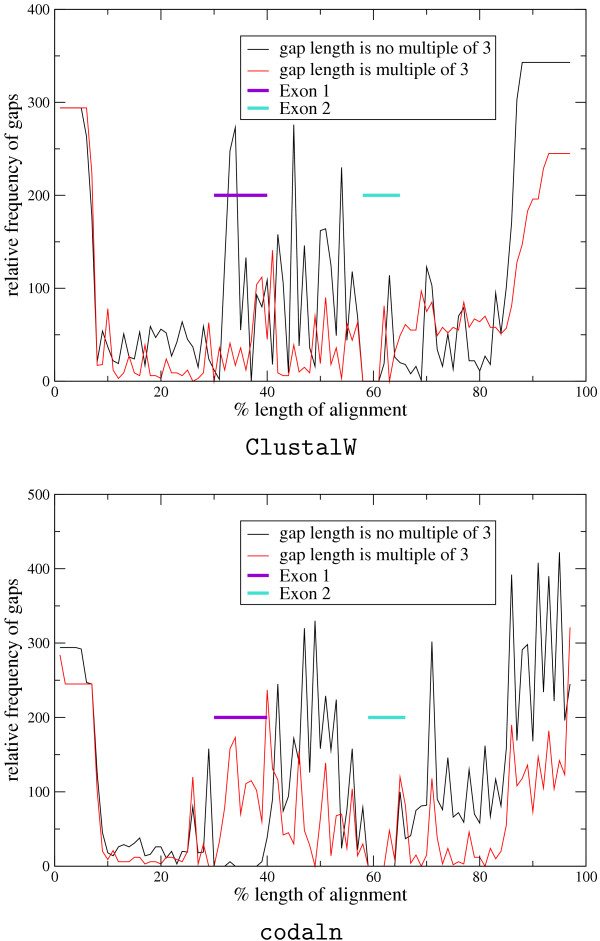
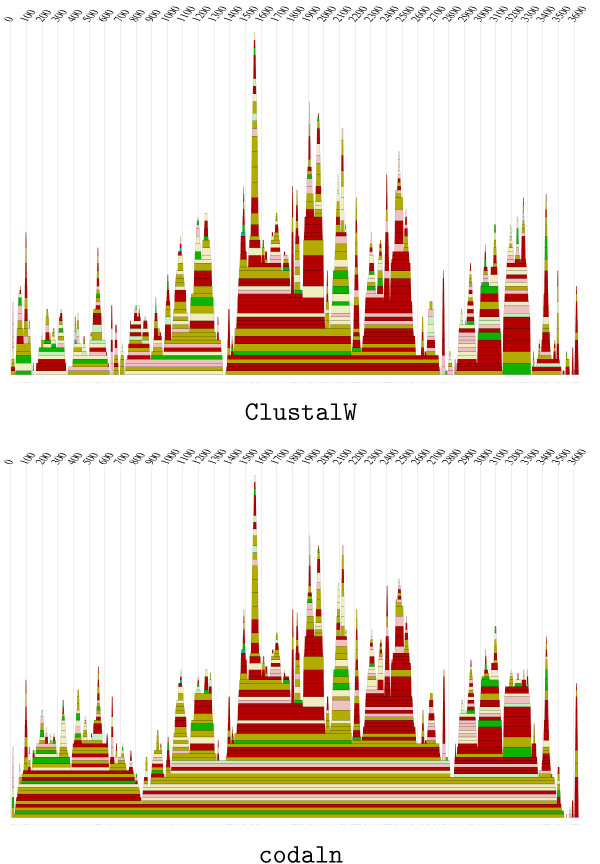
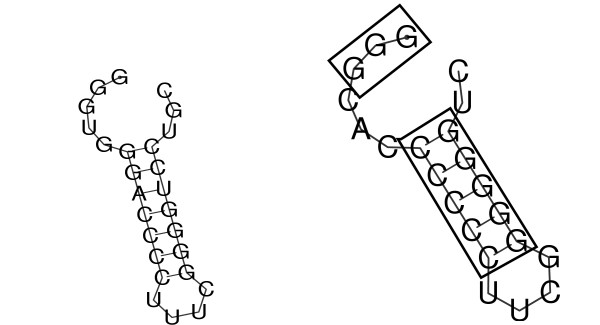
Conclusion: We demonstrate the applicability of codaln to various biologically relevant types of sequences (bacteriophage Levivirus and Vertebrate Hox clusters) and show that the combination of nucleic acid and amino acid sequence information leads to improved alignments. These, in turn, increase the performance of analysis tools that depend strictly on good input alignments such as methods for detecting conserved RNA secondary structure elements.
Figures


Similar articles
-
transAlign: using amino acids to facilitate the multiple alignment of protein-coding DNA sequences.BMC Bioinformatics. 2005 Jun 22;6:156. doi: 10.1186/1471-2105-6-156. BMC Bioinformatics. 2005. PMID: 15969769 Free PMC article.
-
Considerations in the identification of functional RNA structural elements in genomic alignments.BMC Bioinformatics. 2007 Jan 30;8:33. doi: 10.1186/1471-2105-8-33. BMC Bioinformatics. 2007. PMID: 17263882 Free PMC article.
-
CGAT: a comparative genome analysis tool for visualizing alignments in the analysis of complex evolutionary changes between closely related genomes.BMC Bioinformatics. 2006 Oct 24;7:472. doi: 10.1186/1471-2105-7-472. BMC Bioinformatics. 2006. PMID: 17062155 Free PMC article.
-
Computation and analysis of genomic multi-sequence alignments.Annu Rev Genomics Hum Genet. 2007;8:193-213. doi: 10.1146/annurev.genom.8.080706.092300. Annu Rev Genomics Hum Genet. 2007. PMID: 17489682 Review.
-
Finding homologs to nucleic acid or protein sequences using the framesearch program.Curr Protoc Bioinformatics. 2002 Aug;Chapter 3:Unit 3.2. doi: 10.1002/0471250953.bi0302s00. Curr Protoc Bioinformatics. 2002. PMID: 18792937 Review.
Cited by
-
MACSE: Multiple Alignment of Coding SEquences accounting for frameshifts and stop codons.PLoS One. 2011;6(9):e22594. doi: 10.1371/journal.pone.0022594. Epub 2011 Sep 16. PLoS One. 2011. PMID: 21949676 Free PMC article.
-
An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells.Nat Microbiol. 2019 Feb;4(2):280-292. doi: 10.1038/s41564-018-0297-1. Epub 2018 Nov 26. Nat Microbiol. 2019. PMID: 30478287 Free PMC article.
-
A novel ilarvirus protein CP-RT is expressed via stop codon readthrough and suppresses RDR6-dependent RNA silencing.PLoS Pathog. 2024 May 30;20(5):e1012034. doi: 10.1371/journal.ppat.1012034. eCollection 2024 May. PLoS Pathog. 2024. PMID: 38814986 Free PMC article.
-
MAGNOLIA: multiple alignment of protein-coding and structural RNA sequences.Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W14-8. doi: 10.1093/nar/gkn321. Epub 2008 May 30. Nucleic Acids Res. 2008. PMID: 18515348 Free PMC article.
-
HBVRegDB: annotation, comparison, detection and visualization of regulatory elements in hepatitis B virus sequences.Virol J. 2007 Dec 17;4:136. doi: 10.1186/1743-422X-4-136. Virol J. 2007. PMID: 18086305 Free PMC article.
References
-
- Thurner C, Hofacker IL, Stadler PF. Conserved RNA Pseudoknots. In: Giegerich R, Stoye J, editor. Proceedings of the GCB 2004 (Bielefeld), Volume P-53 of GI-Edition: Lecture Notes in Informatics. 2004. pp. 207–216.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
