

Full cyclic coordinate descent: solving the protein loop closure problem in Calpha space
- PMID: 15985178
- PMCID: PMC1192790
- DOI: 10.1186/1471-2105-6-159
Full cyclic coordinate descent: solving the protein loop closure problem in Calpha space
Abstract
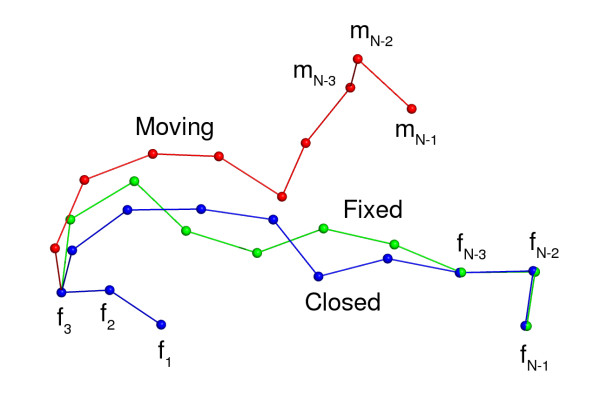
Background: Various forms of the so-called loop closure problem are crucial to protein structure prediction methods. Given an N- and a C-terminal end, the problem consists of finding a suitable segment of a certain length that bridges the ends seamlessly. In homology modelling, the problem arises in predicting loop regions. In de novo protein structure prediction, the problem is encountered when implementing local moves for Markov Chain Monte Carlo simulations. Most loop closure algorithms keep the bond angles fixed or semi-fixed, and only vary the dihedral angles. This is appropriate for a full-atom protein backbone, since the bond angles can be considered as fixed, while the (phi, psi) dihedral angles are variable. However, many de novo structure prediction methods use protein models that only consist of Calpha atoms, or otherwise do not make use of all backbone atoms. These methods require a method that alters both bond and dihedral angles, since the pseudo bond angle between three consecutive Calpha atoms also varies considerably.
Results: Here we present a method that solves the loop closure problem for Calpha only protein models. We developed a variant of Cyclic Coordinate Descent (CCD), an inverse kinematics method from the field of robotics, which was recently applied to the loop closure problem. Since the method alters both bond and dihedral angles, which is equivalent to applying a full rotation matrix, we call our method Full CCD (FCDD). FCCD replaces CCD's vector-based optimization of a rotation around an axis with a singular value decomposition-based optimization of a general rotation matrix. The method is easy to implement and numerically stable.
Conclusion: We tested the method's performance on sets of random protein Calpha segments between 5 and 30 amino acids long, and a number of loops of length 4, 8 and 12. FCCD is fast, has a high success rate and readily generates conformations close to those of real loops. The presence of constraints on the angles only has a small effect on the performance. A reference implementation of FCCD in Python is available as supplementary information.
Figures


Similar articles
-
Cyclic coordinate descent: A robotics algorithm for protein loop closure.Protein Sci. 2003 May;12(5):963-72. doi: 10.1110/ps.0242703. Protein Sci. 2003. PMID: 12717019 Free PMC article.
-
Discrete restraint-based protein modeling and the Calpha-trace problem.Protein Sci. 2003 Sep;12(9):2032-46. doi: 10.1110/ps.0386903. Protein Sci. 2003. PMID: 12931001 Free PMC article.
-
Modeling protein loops using a phi i + 1, psi i dimer database.Protein Sci. 1995 Jul;4(7):1412-20. doi: 10.1002/pro.5560040715. Protein Sci. 1995. PMID: 7670382 Free PMC article.
-
A Review of Protein Structure Prediction Using Lattice Model.Crit Rev Biomed Eng. 2018;46(2):147-162. doi: 10.1615/CritRevBiomedEng.2018026093. Crit Rev Biomed Eng. 2018. PMID: 30055531 Review.
-
Computational methods for high resolution prediction and refinement of protein structures.Curr Opin Struct Biol. 2013 Apr;23(2):177-84. doi: 10.1016/j.sbi.2013.01.010. Epub 2013 May 18. Curr Opin Struct Biol. 2013. PMID: 23688933 Review.
Cited by
-
A self-organizing algorithm for modeling protein loops.PLoS Comput Biol. 2009 Aug;5(8):e1000478. doi: 10.1371/journal.pcbi.1000478. Epub 2009 Aug 21. PLoS Comput Biol. 2009. PMID: 19696883 Free PMC article.
-
Conformational sampling in template-free protein loop structure modeling: an overview.Comput Struct Biotechnol J. 2013 Feb 25;5:e201302003. doi: 10.5936/csbj.201302003. eCollection 2013. Comput Struct Biotechnol J. 2013. PMID: 24688696 Free PMC article. Review.
-
ModeRNA: a tool for comparative modeling of RNA 3D structure.Nucleic Acids Res. 2011 May;39(10):4007-22. doi: 10.1093/nar/gkq1320. Epub 2011 Feb 7. Nucleic Acids Res. 2011. PMID: 21300639 Free PMC article.
-
Improvements to robotics-inspired conformational sampling in rosetta.PLoS One. 2013 May 21;8(5):e63090. doi: 10.1371/journal.pone.0063090. Print 2013. PLoS One. 2013. PMID: 23704889 Free PMC article.
-
Sampling realistic protein conformations using local structural bias.PLoS Comput Biol. 2006 Sep 22;2(9):e131. doi: 10.1371/journal.pcbi.0020131. Epub 2006 Aug 21. PLoS Comput Biol. 2006. PMID: 17002495 Free PMC article.
References
-
- Go N, Scheraga H. Ring closure and local conformational deformations of chain molecules. Macromolecules. 1970;3:178–187. doi: 10.1021/ma60014a012. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
