

Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells
- PMID: 15987447
- PMCID: PMC1175053
- DOI: 10.1186/bcr1019
Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells
Abstract
Introduction: Inhibitors of cyclo-oxygenase (COX)-2 are being extensively studied as anticancer agents. In the present study we evaluated the mechanisms by which a highly selective COX-2 inhibitor, celecoxib, affects tumor growth of two differentially invasive human breast cancer cell lines.
Methods: MDA-MB-231 (highly invasive) and MDA-MB-468 (moderately invasive) cell lines were treated with varying concentrations of celecoxib in vitro, and the effects of this agent on cell growth and angiogenesis were monitored by evaluating cell proliferation, apoptosis, cell cycle arrest, and vasculogenic mimicry. The in vitro results of MDA-MB-231 cell line were further confirmed in vivo in a mouse xenograft model.
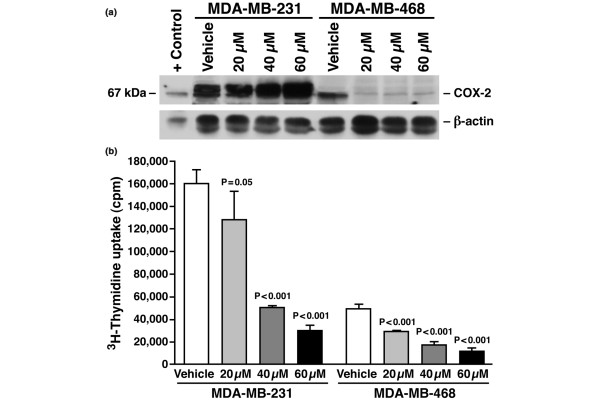
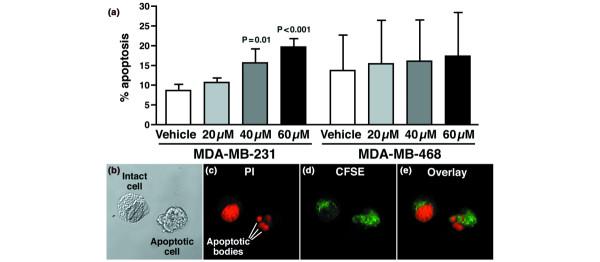
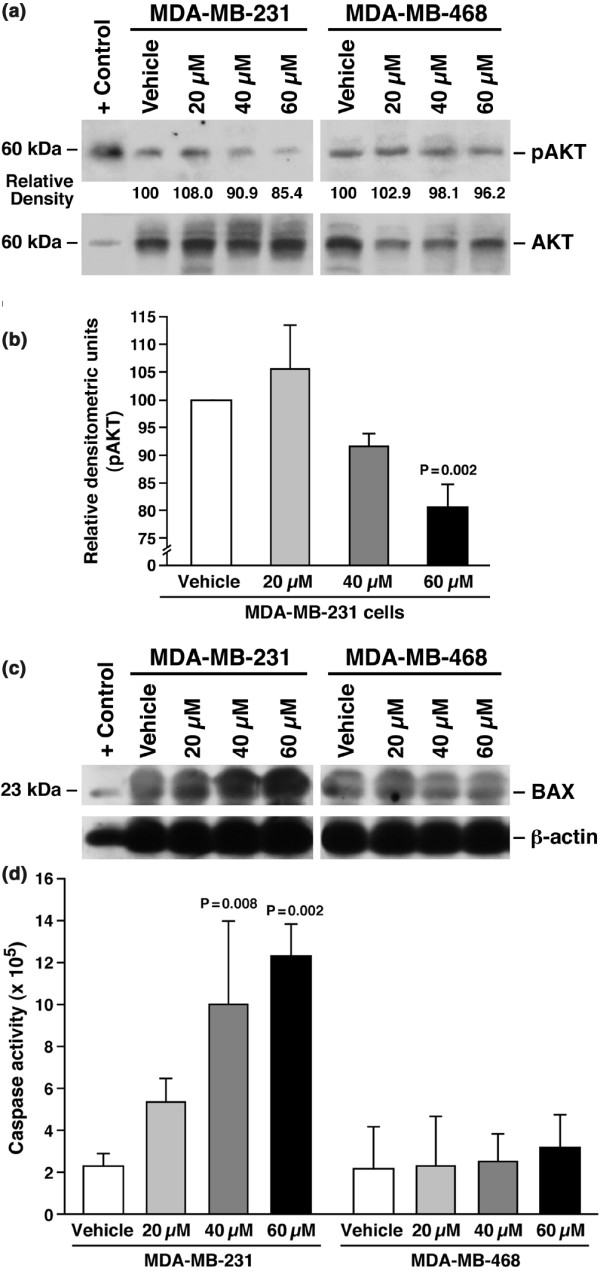
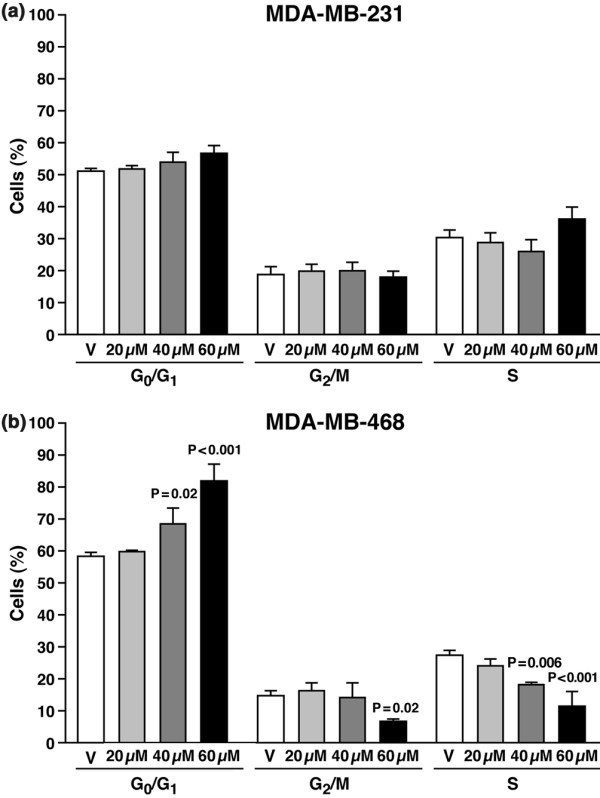
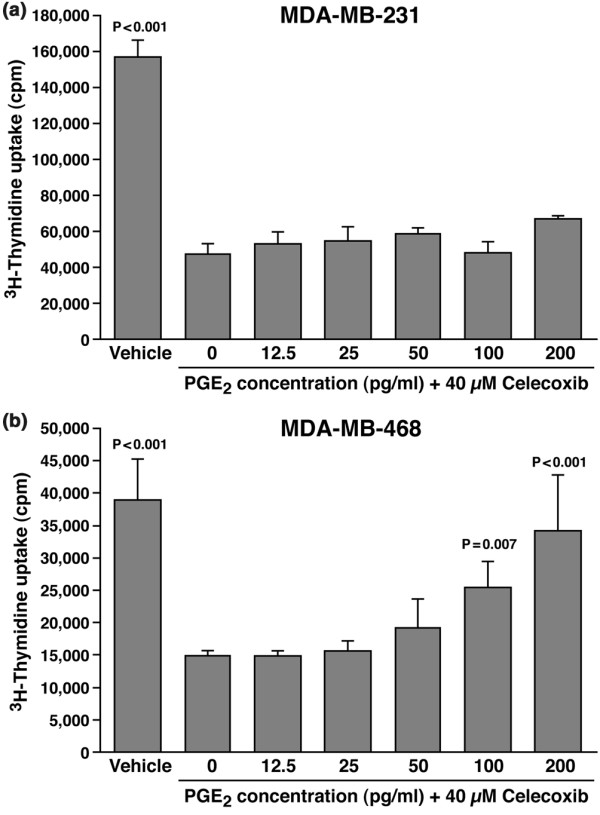
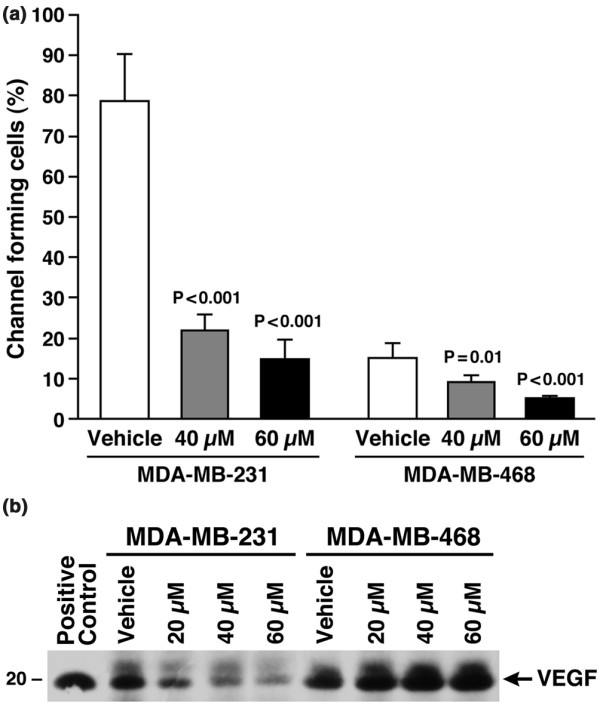
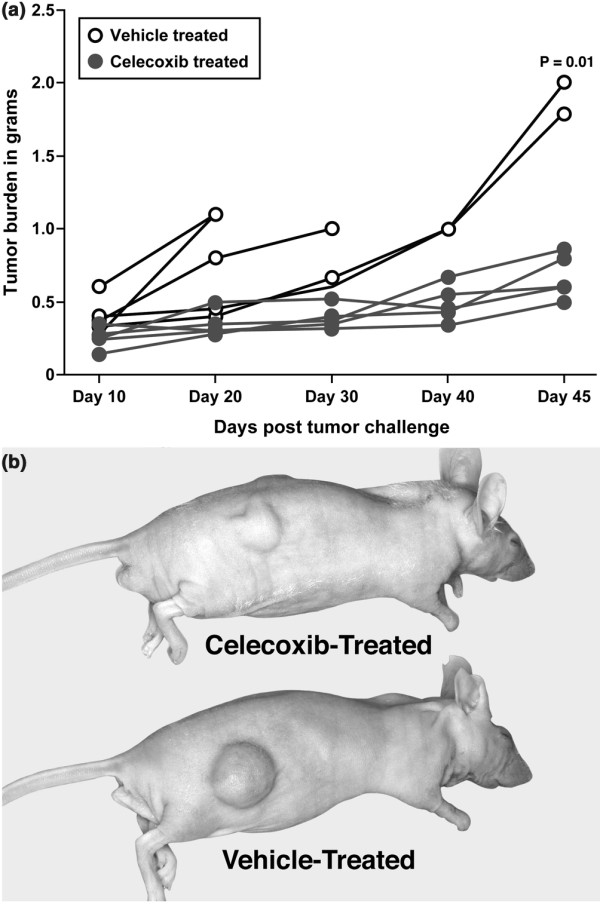
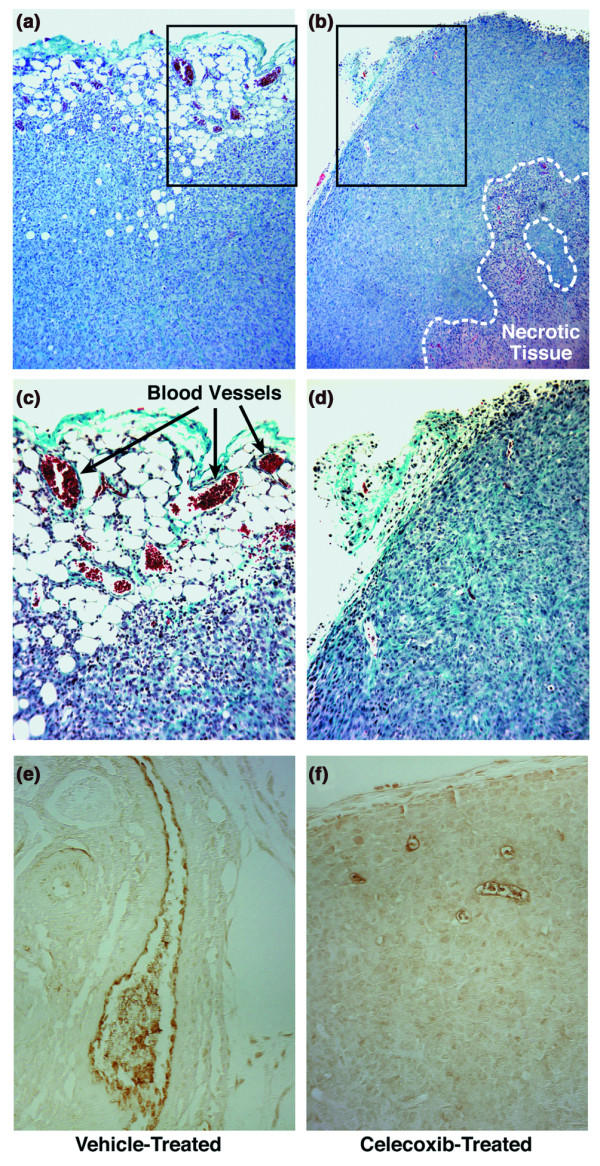
Results: The highly invasive MDA-MB-231 cells express higher levels of COX-2 than do the less invasive MDA-MB-468 cells. Celecoxib treatment inhibited COX-2 activity, indicated by prostaglandin E2 secretion, and caused significant growth arrest in both breast cancer cell lines. In the highly invasive MDA-MB-231 cells, the mechanism of celecoxib-induced growth arrest was by induction of apoptosis, associated with reduced activation of protein kinase B/Akt, and subsequent activation of caspases 3 and 7. In the less invasive MDA-MB-468 cells, growth arrest was a consequence of cell cycle arrest at the G0/G1 checkpoint. Celecoxib-induced growth inhibition was reversed by addition of exogenous prostaglandin E2 in MDA-MB-468 cells but not in MDA-MB-231 cells. Furthermore, MDA-MB-468 cells formed significantly fewer extracellular matrix associated microvascular channels in vitro than did the high COX-2 expressing MDA-MB-231 cells. Celecoxib treatment not only inhibited cell growth and vascular channel formation but also reduced vascular endothelial growth factor levels. The in vitro findings corroborated in vivo data from a mouse xenograft model in which daily administration of celecoxib significantly reduced tumor growth of MDA-MB-231 cells, which was associated with reduced vascularization and increased necrosis in the tumor mass.
Conclusion: The disparate molecular mechanisms of celecoxib-induced growth inhibition in human breast cancer cells depends upon the level of COX-2 expression and the invasive potential of the cell lines examined. Data suggest a role for COX-2 not only in the growth of cancer cells but also in activating the angiogenic pathway through regulating levels of vascular endothelial growth factor.
Figures
References
-
- Harris RE, Chlebowski RT, Jackson RD, Frid DJ, Ascenseo JL, Anderson G, Loar A, Rodabough RJ, White E, McTiernan A. Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women's Health Initiative. Cancer Res. 2003;63:6096–6101. - PubMed
-
- Alshafie GA, Harris RE, Robertson FM, Parrett ML, Ross M, Abou-Issa H. Comparative chemopreventive activity of ibuprofen and N-(4-hydroxyphenyl) retinamide against the development and growth of rat mammary adenocarcinomas. Anticancer Res. 1999;19:3031–3036. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
