

Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination
- PMID: 15994790
- PMCID: PMC1168795
- DOI: 10.1128/JVI.79.14.8960-8968.2005
Low CD8 T-cell proliferative potential and high viral load limit the effectiveness of therapeutic vaccination
Abstract
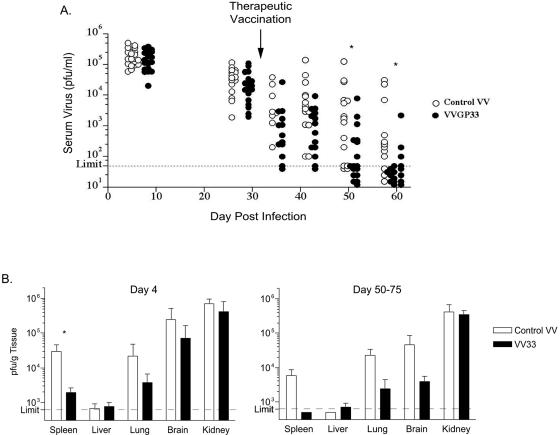
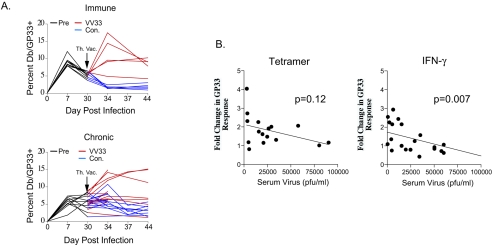
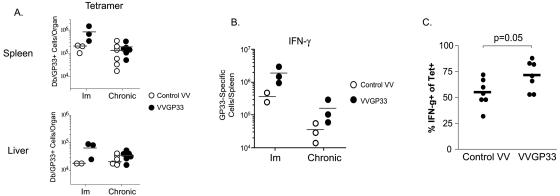
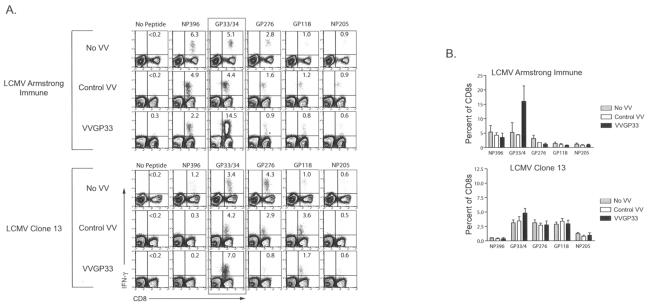
Therapeutic vaccination has the potential to boost immune responses and enhance viral control during chronic infections. However, many therapeutic vaccination approaches have fallen short of expectations, and effective boosting of antiviral T-cell responses is not always observed. To examine these issues, we studied the impact of therapeutic vaccination, using a murine model of chronic infection with lymphocytic choriomeningitis virus (LCMV). Our results demonstrate that therapeutic vaccination using a recombinant vaccinia virus expressing the LCMV GP33 CD8 T-cell epitope can be effective at accelerating viral control. However, mice with lower viral loads at the time of vaccination responded better to therapeutic vaccination than did those with high viral loads. Also, the proliferative potential of GP33-specific CD8 T cells from chronically infected mice was substantially lower than that of GP33-specific memory CD8 T cells from mice with immunity to LCMV, suggesting that poor T-cell expansion may be an important reason for suboptimal responses to therapeutic vaccination. Thus, our results highlight the potential positive effects of therapeutic vaccination on viral control during chronic infection but also provide evidence that a high viral load at the time of vaccination and the low proliferative potential of responding T cells are likely to limit the effectiveness of therapeutic vaccination.
Figures
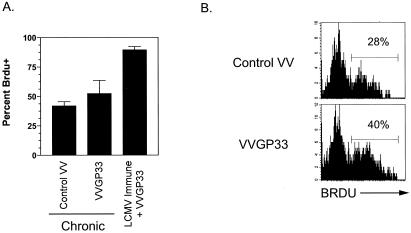
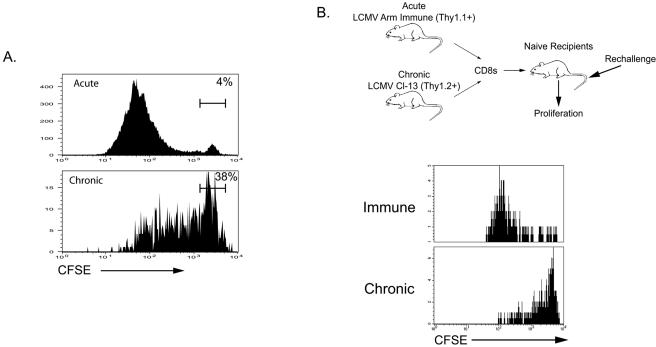
References
-
- Ahmed, R., A. Salmi, L. D. Butler, J. M. Chiller, and M. B. Oldstone. 1984. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J. Exp. Med. 160:521-540. - PMC - PubMed
-
- Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407:386-390. - PubMed
-
- Autran, B., G. Carcelain, B. Combadiere, and P. Debre. 2004. Therapeutic vaccines for chronic infections. Science 305:205-208. - PubMed
-
- Blattman, J. N., J. M. Grayson, E. J. Wherry, S. M. Kaech, K. A. Smith, and R. Ahmed. 2003. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat. Med. 9:540-547. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
