

Suppressors of a host range mutation in the rabbitpox virus serpin SPI-1 map to proteins essential for viral DNA replication
- PMID: 15994811
- PMCID: PMC1168772
- DOI: 10.1128/JVI.79.14.9168-9179.2005
Suppressors of a host range mutation in the rabbitpox virus serpin SPI-1 map to proteins essential for viral DNA replication
Abstract
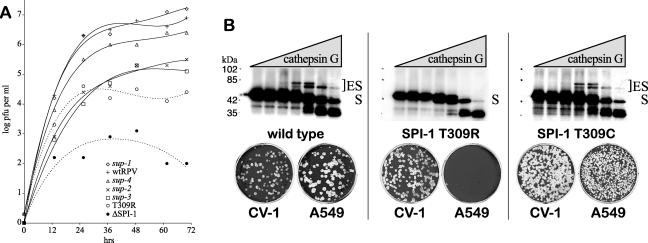
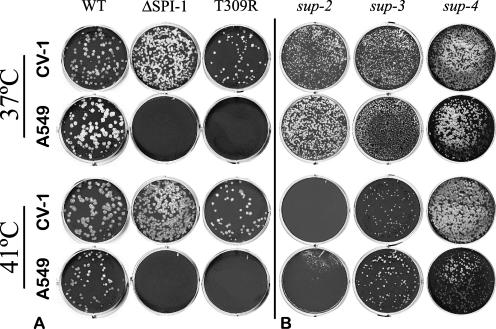
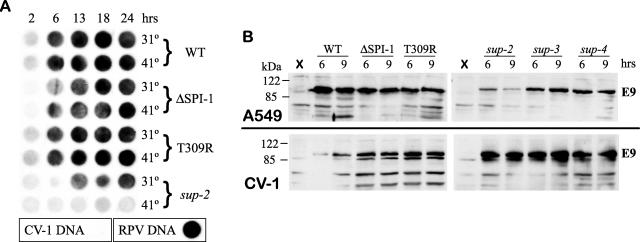
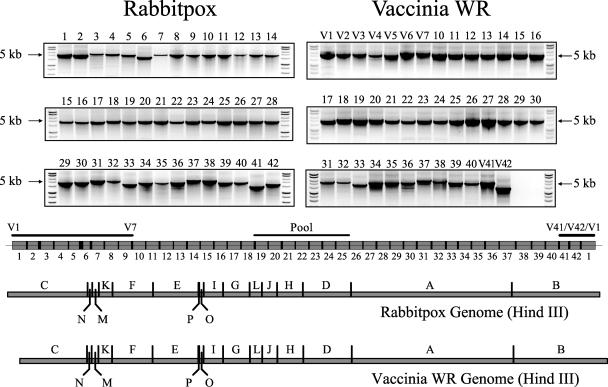
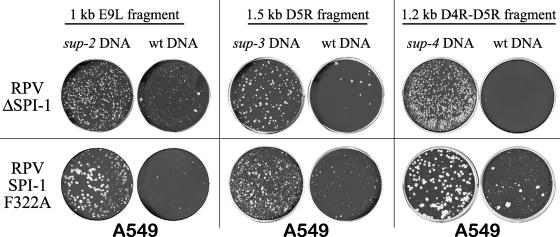
The orthopoxvirus serpin SPI-1 is an intracellular serine protease inhibitor that is active against cathepsin G in vitro. Rabbitpox virus (RPV) mutants with deletions of the SPI-1 gene grow on monkey kidney cells (CV-1) but do not plaque on normally permissive human lung carcinoma cells (A549). This reduced-host-range (hr) phenotype suggests that SPI-1 may interact with cellular and/or other viral proteins. We devised a genetic screen for suppressors of SPI-1 hr mutations by first introducing a mutation into SPI-1 (T309R) at residue P14 of the serpin reactive center loop. The SPI-1 T309R serpin is inactive as a protease inhibitor in vitro. Introduction of the mutation into RPV leads to the same restricted hr phenotype as deletion of the SPI-1 gene. Second-site suppressors were selected by restoration of growth of the RPV SPI-1 T309R hr mutant on A549 cells. Both intragenic and extragenic suppressors of the T309R mutation were identified. One novel intragenic suppressor mutation, T309C, restored protease inhibition by SPI-1 in vitro. Extragenic suppressor mutations were mapped by a new procedure utilizing overlapping PCR products encompassing the entire genome in conjunction with marker rescue. One suppressor mutation, which also rendered the virus temperature sensitive for growth, mapped to the DNA polymerase gene (E9L). Several other suppressors mapped to gene D5R, an NTPase required for DNA replication. These results unexpectedly suggest that the host range function of SPI-1 may be associated with viral DNA replication by an as yet unknown mechanism.
Figures
Similar articles
-
A rabbitpox virus serpin gene controls host range by inhibiting apoptosis in restrictive cells.J Virol. 1995 Dec;69(12):7688-98. doi: 10.1128/JVI.69.12.7688-7698.1995. J Virol. 1995. PMID: 7494278 Free PMC article.
-
The SPI-1 gene of rabbitpox virus determines host range and is required for hemorrhagic pock formation.Virology. 1994 Jul;202(1):305-14. doi: 10.1006/viro.1994.1347. Virology. 1994. PMID: 8009842
-
Vaccinia virus serpin-1 deletion mutant exhibits a host range defect characterized by low levels of intermediate and late mRNAs.Virology. 1999 Sep 30;262(2):298-311. doi: 10.1006/viro.1999.9884. Virology. 1999. PMID: 10502509
-
Vaccinia virus DNA replication: a short review.Biochimie. 1995;77(10):774-9. doi: 10.1016/0300-9084(96)88195-8. Biochimie. 1995. PMID: 8824774 Review.
-
The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses.Virology. 1998 May 10;244(2):365-96. doi: 10.1006/viro.1998.9123. Virology. 1998. PMID: 9601507 Review.
Cited by
-
The vaccinia virus E8R gene product is required for formation of transcriptionally active virions.Virology. 2007 Oct 25;367(2):398-412. doi: 10.1016/j.virol.2007.05.002. Epub 2007 Jul 9. Virology. 2007. PMID: 17619043 Free PMC article.
-
Marker rescue mapping of the combined Condit/Dales collection of temperature-sensitive vaccinia virus mutants.Virology. 2008 May 25;375(1):213-22. doi: 10.1016/j.virol.2008.01.027. Epub 2008 Mar 7. Virology. 2008. PMID: 18314155 Free PMC article.
-
Isolation and characterization of cidofovir resistant vaccinia viruses.Virol J. 2008 May 14;5:58. doi: 10.1186/1743-422X-5-58. Virol J. 2008. PMID: 18479513 Free PMC article.
-
Vaccinia virus temperature-sensitive mutants in the A28 gene produce non-infectious virions that bind to cells but are defective in entry.Virology. 2007 Sep 15;366(1):62-72. doi: 10.1016/j.virol.2007.03.060. Epub 2007 May 17. Virology. 2007. PMID: 17499330 Free PMC article.
-
Mapping and phenotypic analysis of spontaneous isatin-beta-thiosemicarbazone resistant mutants of vaccinia virus.Virology. 2007 Jul 5;363(2):319-32. doi: 10.1016/j.virol.2007.02.005. Epub 2007 Mar 1. Virology. 2007. PMID: 17336362 Free PMC article.
References
-
- Ali, A. N., P. C. Turner, M. A. Brooks, and R. W. Moyer. 1994. The SPI-1 gene of rabbitpox virus determines host range and is required for hemorrhagic pock formation. Virology 202:306-314. - PubMed
-
- Anderson, C. W., M. M. Hardy, J. J. Dunn, and D. F. Klessig. 1983. Independent, spontaneous mutants of adenovirus type 2-simian virus 40 hybrid Ad2+ ND3 that grow efficiently in monkey cells possess indentical [sic] mutations in the adenovirus type 2 DNA-binding protein gene. J. Virol. 48:31-39. - PMC - PubMed
-
- Becerra, S. P., A. Sagasti, P. Spinella, and V. Notario. 1995. Pigment epithelium-derived factor behaves like a noninhibitory serpin. Neurotrophic activity does not require the serpin reactive loop. J. Biol. Chem. 270:25992-25999. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
