

Alms1-disrupted mice recapitulate human Alström syndrome
- PMID: 16000322
- PMCID: PMC2862911
- DOI: 10.1093/hmg/ddi235
Alms1-disrupted mice recapitulate human Alström syndrome
Abstract
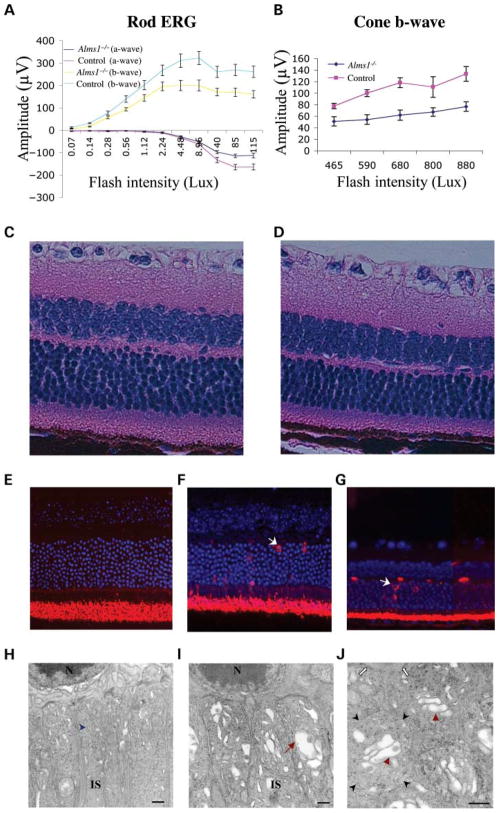
Mutations in the human ALMS1 gene cause Alström syndrome (AS), a progressive disease characterized by neurosensory deficits and by metabolic defects including childhood obesity, hyperinsulinemia and Type 2 diabetes. Other features that are more variable in expressivity include dilated cardiomyopathy, hypertriglyceridemia, hypercholesterolemia, scoliosis, developmental delay and pulmonary and urological dysfunctions. ALMS1 encodes a ubiquitously expressed protein of unknown function. To obtain an animal model in which the etiology of the observed pathologies could be further studied, we generated a mouse model using an Alms1 gene-trapped ES cell line. Alms1-/- mice develop features similar to patients with AS, including obesity, hypogonadism, hyperinsulinemia, retinal dysfunction and late-onset hearing loss. Insulin resistance and increased body weight are apparent between 8 and 12 weeks of age, with hyperglycemia manifesting at approximately 16 weeks of age. In addition, Alms1-/- mice have normal hearing until 8 months of age, after which they display abnormal auditory brainstem responses. Diminished cone ERG b-wave response is observed early, followed by the degeneration of photoreceptor cells. Electron microscopy revealed accumulation of intracellular vesicles in the inner segments of photoreceptors, whereas immunohistochemical analysis showed mislocalization of rhodopsin to the outer nuclear layer. These findings suggest that ALMS1 has a role in intracellular trafficking.
Conflict of interest statement
Figures






References
-
- Marshall JD, Ludman MD, Shea SE, Salisbury SR, Willi SM, LaRoche RG, Nishina PM. Genealogy, natural history, and phenotype of Alstrom syndrome in a large Acadian kindred and three additional families. Am J Med Genet. 1997;73:150–161. - PubMed
-
- Michaud JL, Heon E, Guilbert F, Weill J, Puech B, Benson L, Smallhorn JF, Shuman CT, Buncic JR, Levin AV, et al. Natural history of Alstrom syndrome in early childhood: onset with dilated cardiomyopathy. J Pediatr. 1996;128:225–229. - PubMed
-
- Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence–Moon–Bardet–Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand. 1959;34:1–35. - PubMed
-
- Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, et al. New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med. 2005;165:675–683. - PubMed
-
- Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, et al. Mutations in ALMS1 cause obesity, Type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
