

Genome-wide scans for loci under selection in humans
- PMID: 16004726
- PMCID: PMC3525256
- DOI: 10.1186/1479-7364-2-2-113
Genome-wide scans for loci under selection in humans
Abstract
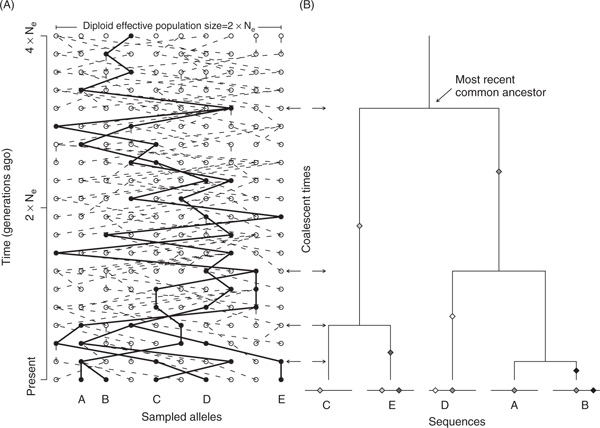
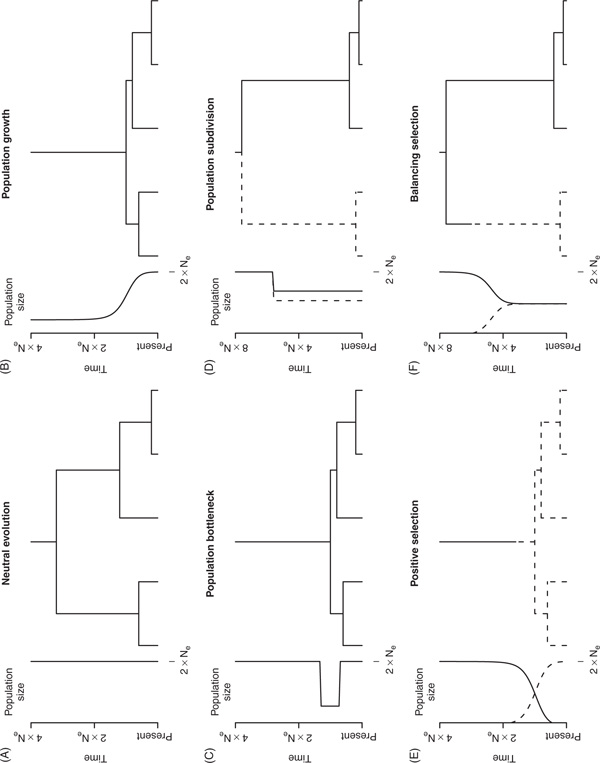
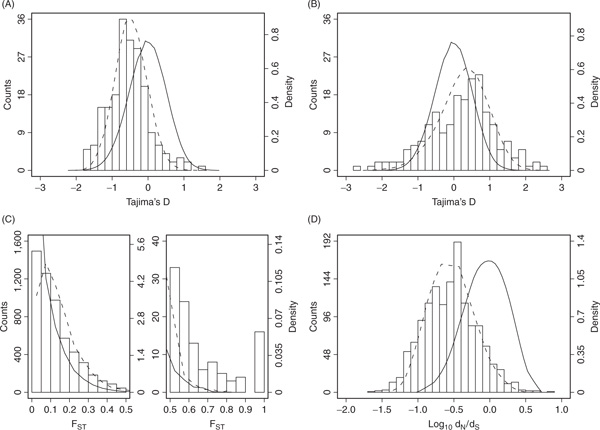
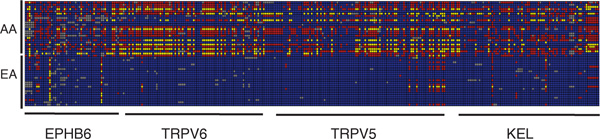
Natural selection, which can be defined as the differential contribution of genetic variants to future generations, is the driving force of Darwinian evolution. Identifying regions of the human genome that have been targets of natural selection is an important step in clarifying human evolutionary history and understanding how genetic variation results in phenotypic diversity, it may also facilitate the search for complex disease genes. Technological advances in high-throughput DNA sequencing and single nucleotide polymorphism genotyping have enabled several genome-wide scans of natural selection to be undertaken. Here, some of the observations that are beginning to emerge from these studies will be reviewed, including evidence for geographically restricted selective pressures (ie local adaptation) and a relationship between genes subject to natural selection and human disease. In addition, the paper will highlight several important problems that need to be addressed in future genome-wide studies of natural selection.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
