

Tachpyridine, a metal chelator, induces G2 cell-cycle arrest, activates checkpoint kinases, and sensitizes cells to ionizing radiation
- PMID: 16014567
- PMCID: PMC1895322
- DOI: 10.1182/blood-2005-03-1263
Tachpyridine, a metal chelator, induces G2 cell-cycle arrest, activates checkpoint kinases, and sensitizes cells to ionizing radiation
Abstract
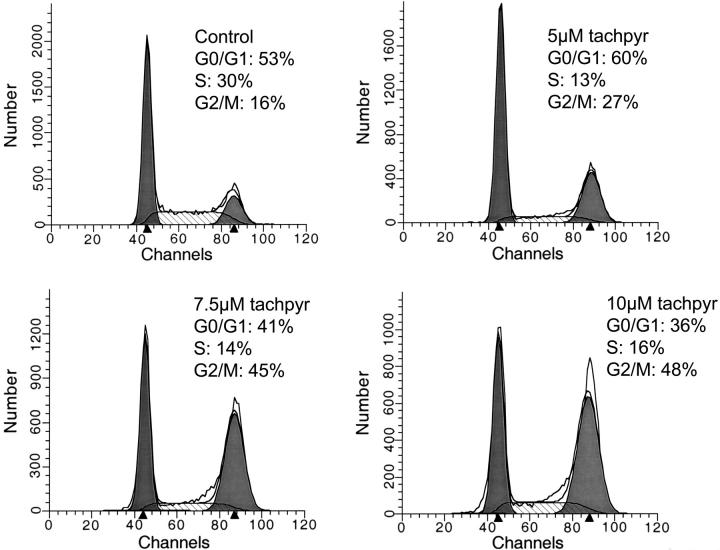
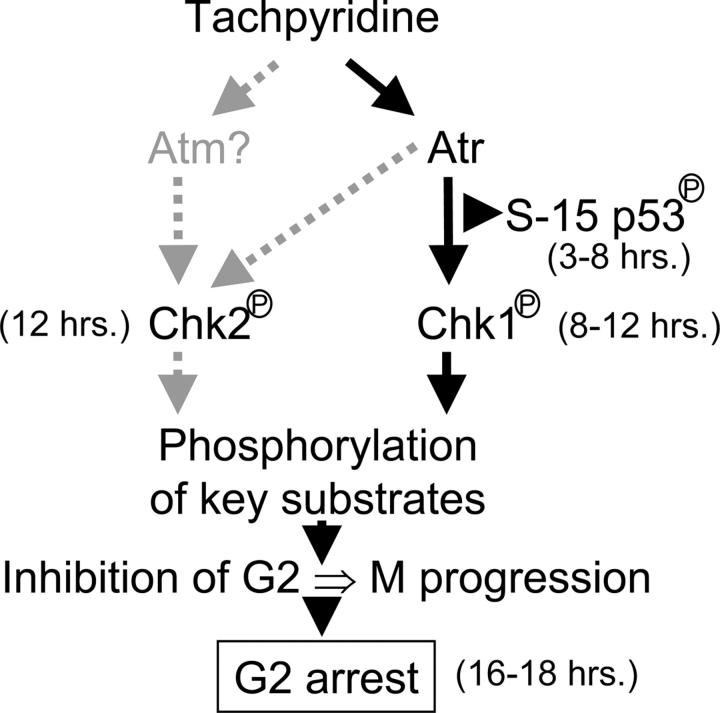
Iron is critical for cell growth and proliferation. Iron chelators are being explored for a number of clinical applications, including the treatment of neurodegenerative disorders, heart disease, and cancer. To uncover mechanisms of action of tachpyridine, a chelator currently undergoing preclinical evaluation as an anticancer agent, cell-cycle analysis was performed. Tachpyridine arrested cells at G2, a radiosensitive phase of the cell cycle, and enhanced the sensitivity of cancer cells but not nontransformed cells to ionizing radiation. G2 arrest was p53 independent and was accompanied by activation of the checkpoint kinases CHK1 and CHK2. G2 arrest was blocked by UCN-01, a CHK1 inhibitor, but proceeded in CHK2 knock-out cells, indicating a critical role for CHK1 in G2 arrest. Tachpyridine-induced cell-cycle arrest was abrogated in cells treated with caffeine, an inhibitor of the ataxia-telangiectasia mutated/ataxia-telangiectasia-mutated and Rad3-related (ATM/ATR) kinases. Further, G2 arrest proceeded in ATM-deficient cells but was blocked in ATR-deficient cells, implicating ATR as the proximal kinase in tachpyridine-mediated G2 arrest. Collectively, our results suggest that iron chelators may function as antitumor and radioenhancing agents and uncover a previously unexplored activity of iron chelators in activation of ATR and checkpoint kinases.
Figures





Similar articles
-
Cadmium-induced DNA damage triggers G(2)/M arrest via chk1/2 and cdc2 in p53-deficient kidney proximal tubule cells.Am J Physiol Renal Physiol. 2010 Feb;298(2):F255-65. doi: 10.1152/ajprenal.00273.2009. Epub 2009 Nov 18. Am J Physiol Renal Physiol. 2010. PMID: 19923412
-
Irradiation-induced G2/M checkpoint response requires ERK1/2 activation.Oncogene. 2007 Jul 12;26(32):4689-98. doi: 10.1038/sj.onc.1210268. Epub 2007 Feb 5. Oncogene. 2007. PMID: 17297454
-
Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional.Oncogene. 2011 Oct 13;30(41):4261-74. doi: 10.1038/onc.2011.135. Epub 2011 May 2. Oncogene. 2011. PMID: 21532626
-
Novel regulation of checkpoint kinase 1: Is checkpoint kinase 1 a good candidate for anti-cancer therapy?Cancer Sci. 2012 Jul;103(7):1195-200. doi: 10.1111/j.1349-7006.2012.02280.x. Epub 2012 Apr 23. Cancer Sci. 2012. PMID: 22435685 Free PMC article. Review.
-
An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells.Cell Cycle. 2007 Jul 15;6(14):1682-6. doi: 10.4161/cc.6.14.4480. Epub 2007 May 21. Cell Cycle. 2007. PMID: 17637566 Review.
Cited by
-
The potential effectiveness of nanoparticles as radio sensitizers for radiotherapy.Bioimpacts. 2014;4(1):15-20. doi: 10.5681/bi.2014.003. Epub 2014 Mar 8. Bioimpacts. 2014. PMID: 24790894 Free PMC article. Review.
-
Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells.Int J Mol Sci. 2020 Oct 16;21(20):7674. doi: 10.3390/ijms21207674. Int J Mol Sci. 2020. PMID: 33081324 Free PMC article.
-
Transferrin Receptor-Mediated Iron Uptake Promotes Colon Tumorigenesis.Adv Sci (Weinh). 2023 Apr;10(10):e2207693. doi: 10.1002/advs.202207693. Epub 2023 Jan 26. Adv Sci (Weinh). 2023. PMID: 36703617 Free PMC article.
-
A three-in-one-bullet for oesophageal cancer: replication fork collapse, spindle attachment failure and enhanced radiosensitivity generated by a ruthenium(ii) metallo-intercalator.Chem Sci. 2017 Nov 16;9(4):841-849. doi: 10.1039/c7sc03712k. eCollection 2018 Jan 28. Chem Sci. 2017. PMID: 29629151 Free PMC article.
-
Revised model for cell cycle regulation by iron: differential roles between transferrin and ferritin.Redox Biol. 2025 Sep;85:103727. doi: 10.1016/j.redox.2025.103727. Epub 2025 Jun 11. Redox Biol. 2025. PMID: 40527018 Free PMC article.
References
-
- Faulk WP, Hsi BL, Stevens PJ. Transferrin and transferrin receptors in carcinoma of the breast. Lancet. 1980;2: 390-392. - PubMed
-
- Seymour GJ, Walsh MD, Lavin MF, Strutton G, Gardiner RA. Transferrin receptor expression by human bladder transitional cell carcinomas. Urol Res. 1987;15: 341-344. - PubMed
-
- Richardson DR, Tran EH, Ponka P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995;86: 4295-4306. - PubMed
-
- Richardson DR, Milnes K. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents II: the mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone. Blood. 1997;89: 3025-3038. - PubMed
-
- Richardson DR. Potential of iron chelators as effective antiproliferative agents. Can J Physiol Pharmacol. 1997;75: 1164-1180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
