

Regulation of p53-MDMX interaction by casein kinase 1 alpha
- PMID: 16024788
- PMCID: PMC1190343
- DOI: 10.1128/MCB.25.15.6509-6520.2005
Regulation of p53-MDMX interaction by casein kinase 1 alpha
Abstract
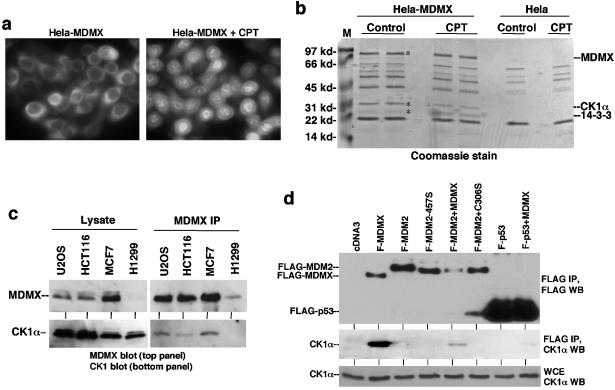
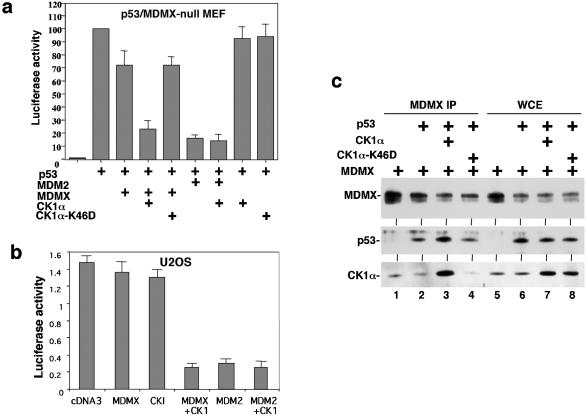
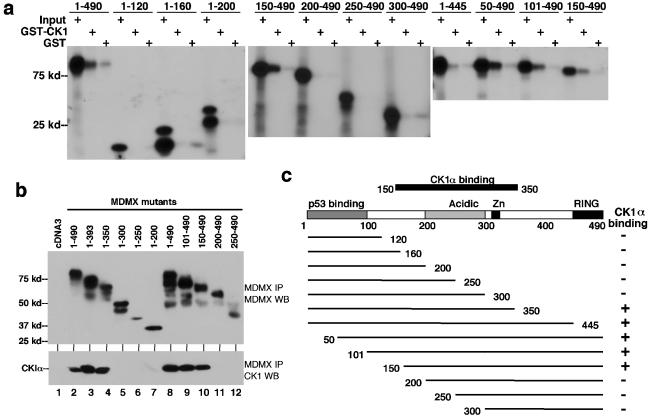
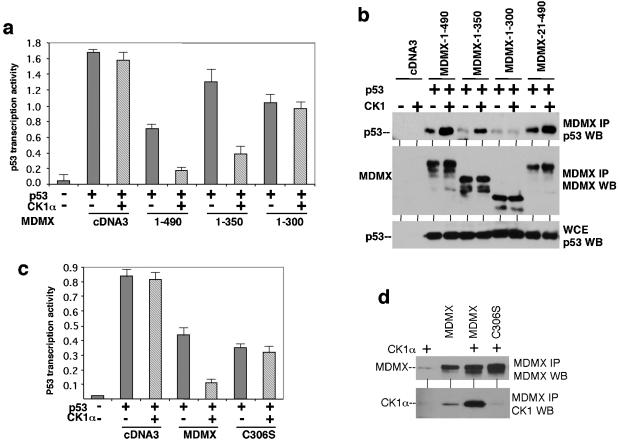
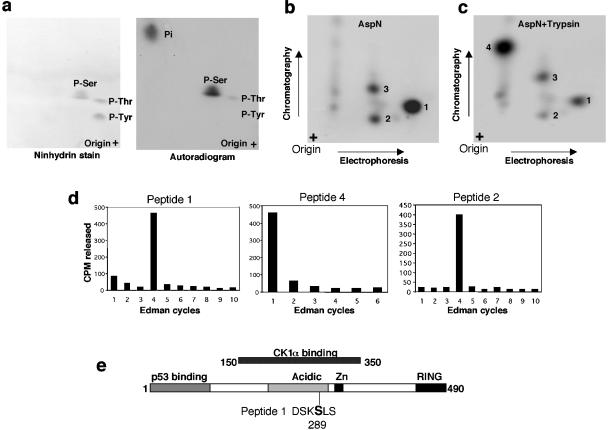
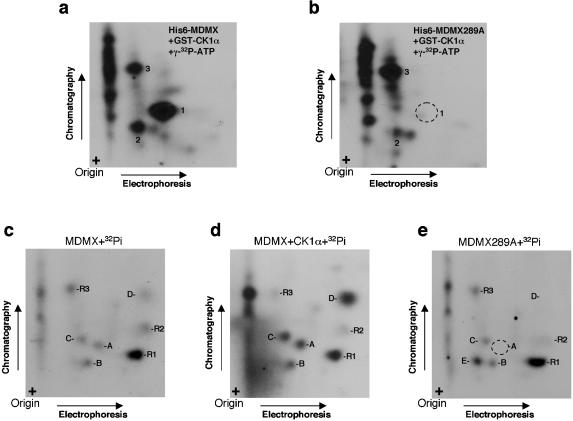
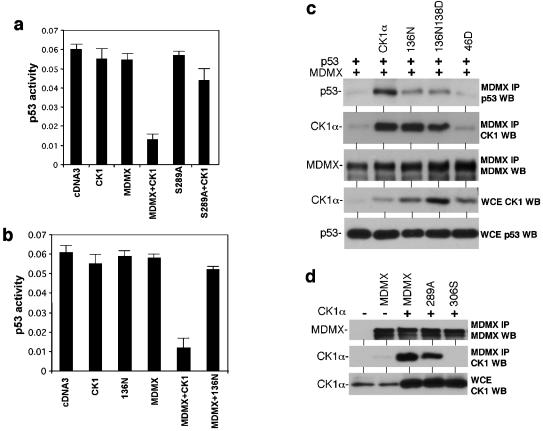
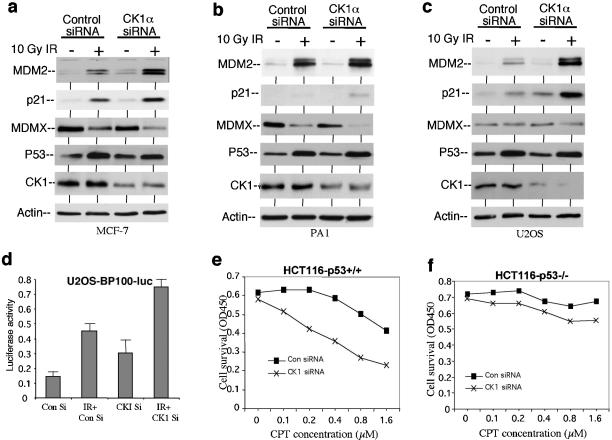
MDMX is a homolog of MDM2 that is critical for regulating p53 function during mouse development. MDMX degradation is regulated by MDM2-mediated ubiquitination. Whether there are other mechanisms of MDMX regulation is largely unknown. We found that MDMX binds to the casein kinase 1 alpha isoform (CK1alpha) and is phosphorylated by CK1alpha. Expression of CK1alpha stimulates the ability of MDMX to bind to p53 and inhibit p53 transcriptional function. Regulation of MDMX-p53 interaction requires CK1alpha binding to the central region of MDMX and phosphorylation of MDMX on serine 289. Inhibition of CK1alpha expression by isoform-specific small interfering RNA (siRNA) activates p53 and further enhances p53 activity after ionizing irradiation. CK1alpha siRNA also cooperates with DNA damage to induce apoptosis. These results suggest that CK1alpha is a functionally relevant MDMX-binding protein and plays an important role in regulating p53 activity in the absence or presence of stress.
Figures

References
-
- Baker, S. J., S. Markowitz, E. R. Fearon, J. K. Willson, and B. Vogelstein. 1990. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 249:912-915. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous