

Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth
- PMID: 16036105
- PMCID: PMC1501283
- DOI: 10.1593/neo.04571
Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth
Abstract
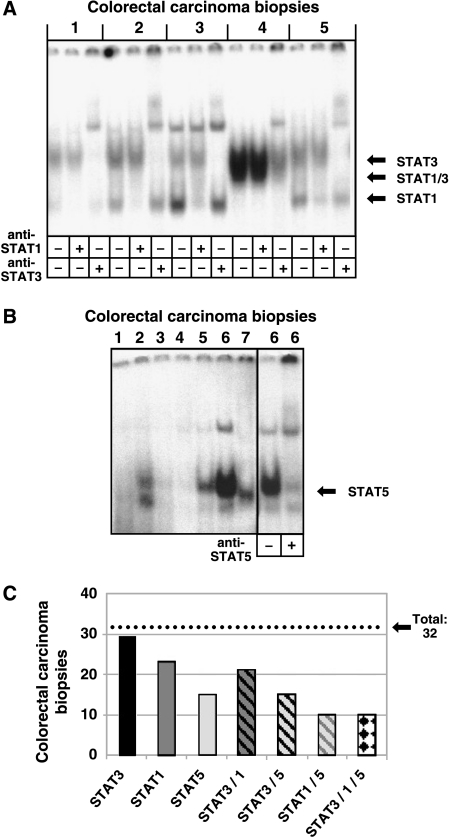
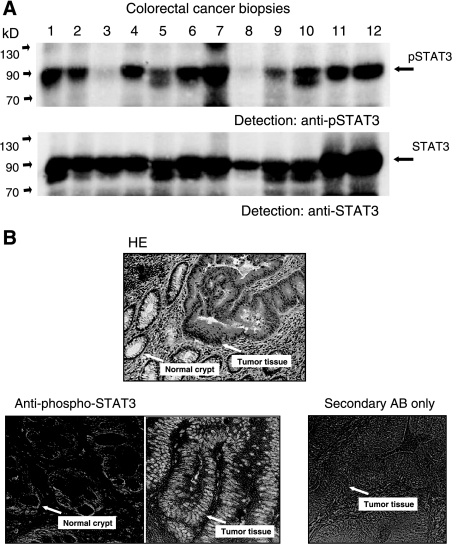
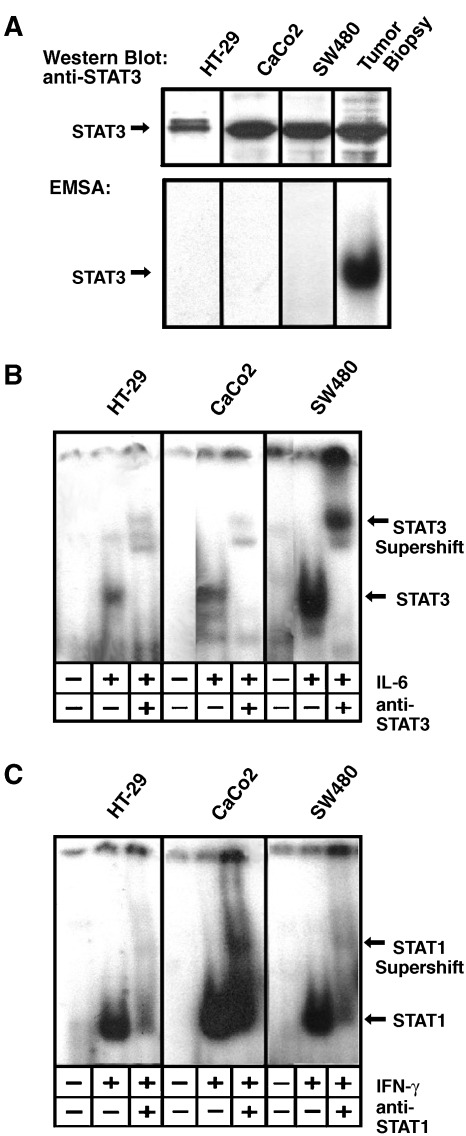
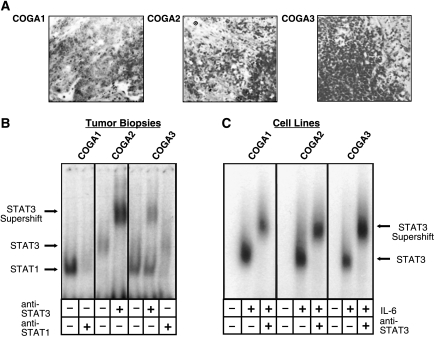
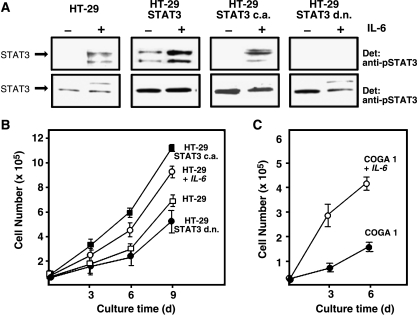
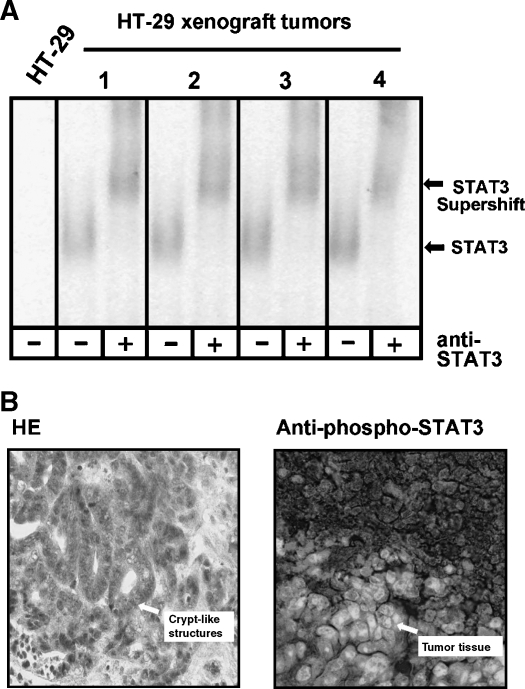
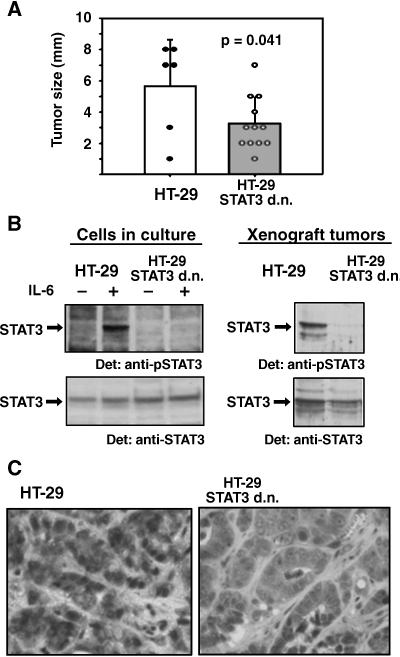
Colorectal carcinoma (CRC) is a major cause of morbidity and mortality in Western countries. It has so far been molecularly defined mainly by alterations of the Wnt pathway. We show here for the first time that aberrant activities of the signal transducer and activator of transcription STAT3 actively contribute to this malignancy and, thus, are a potential therapeutic target for CRC. Constitutive STAT3 activity was found to be abundant in dedifferentiated cancer cells and infiltrating lymphocytes of CRC samples, but not in non-neoplastic colon epithelium. Cell lines derived from malignant colorectal tumors lost persistent STAT3 activity in culture. However, implantation of colon carcinoma cells into nude mice resulted in restoration of STAT3 activity, suggesting a role of an extracellular stimulus within the tumor microenvironment as a trigger for STAT activation. STAT3 activity in CRC cells triggered through interleukin-6 or through a constitutively active STAT3 mutant promoted cancer cell multiplication, whereas STAT3 inhibition through a dominant-negative variant impaired IL-6-driven proliferation. Blockade of STAT3 activation in CRC-derived xenograft tumors slowed down their development, arguing for a contribution of STAT3 to colorectal tumor growth.
Figures
References
-
- Declan Fleming RY. Colorectal cancer screening and follow-up. Surg Oncol. 1998;7:125–137. - PubMed
-
- Oving IM, Clevers HC. Molecular causes of colon cancer. Eur J Clin Invest. 2002;32:448–457. - PubMed
-
- Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. - PubMed
-
- Ma AH, Xia L, Littman SJ, Swinler S, Lader G, Polinkovsky A, Olechnowicz J, Kasturi L, Lutterbaugh J, Modrich P, et al. Somatic mutation of hPMS2 as a possible cause of sporadic human colon cancer with microsatellite instability. Oncogene. 2000;19:2249–2256. - PubMed
-
- Duval A, Hamelin R. Genetic instability in human mismatch repair deficient cancers. Ann Genet. 2002;45:71–75. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous