

Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria
- PMID: 16043102
- PMCID: PMC2694052
- DOI: 10.1016/j.ymthe.2005.03.025
Low therapeutic threshold for hepatocyte replacement in murine phenylketonuria
Abstract
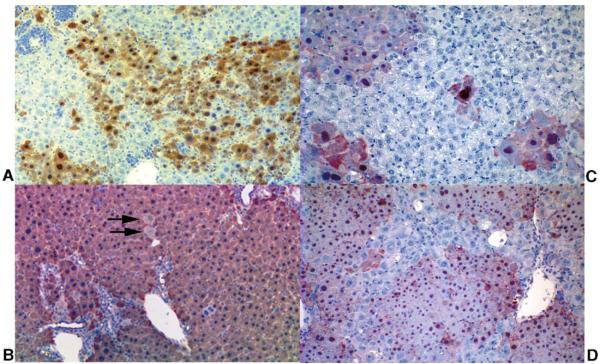
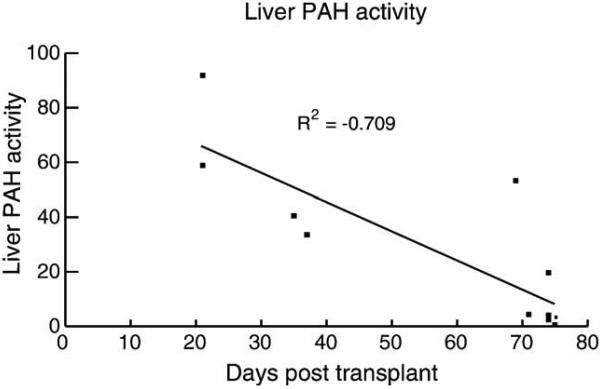
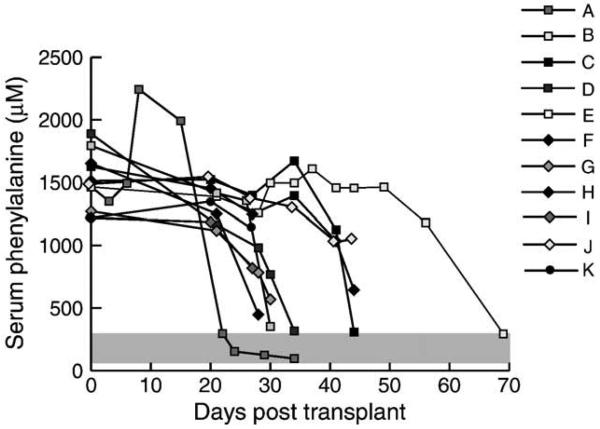
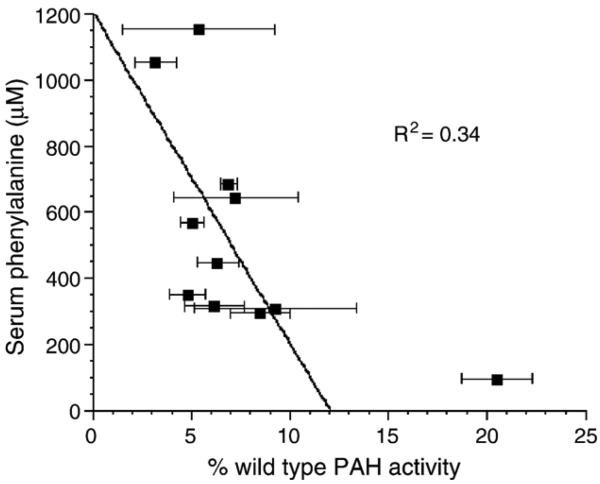
Phenylalanine homeostasis in mammals is primarily controlled by liver phenylalanine hydroxylase (PAH) activity. Inherited PAH deficiency (phenylketonuria or PKU) leads to hyperphenylalaninemia in both mice and humans. A low level of residual liver PAH activity ensures near-normal dietary protein tolerance with normal serum phenylalanine level, but the precise threshold for normal phenylalanine clearance is unknown. We employed hepatocyte transplantation under selective growth conditions to investigate the minimal number of PAH-expressing hepatocytes necessary to prevent hyperphenylalaninemia in mice. Serum phenylalanine levels remained normal in mice exhibiting nearly complete liver repopulation with PAH-deficient hepatocytes (<5% residual wild-type liver PAH activity). Conversely, transplantation of PAH-positive hepatocytes into PAH-deficient Pah(enu2) mice, a model of human PKU, yielded a significant decrease in serum phenylalanine (<700 muM) when liver repopulation exceeded approximately 5%. These data suggest that restoration of phenylalanine homeostasis requires PAH activity in only a minority of hepatocytes.
Figures
References
-
- Scriver CR, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Valle D, editor. The Metabolic & Molecular Bases of Inherited Disease. McGraw–Hill; New York: 2001. pp. 1667–1724.
-
- Udenfriend S, Cooper JR. The enzymatic conversion of phenylalanine to tyrosine. J. Biol. Chem. 1952;194:503–511. - PubMed
-
- Azen CG, et al. Intellectual development in 12-year-old children treated for phenylketonuria. Am. J. Dis. Child. 1991;145:35–39. - PubMed
-
- Fang B, et al. Gene therapy for phenylketonuria: phenotypic correction in a genetically deficient mouse model by adenovirus-mediated hepatic gene therapy. Gene Ther. 1994;1:247–254. - PubMed
-
- Laconi E, Laconi S. Principles of hepatocyte repopulation. Semin. Cell Dev. Biol. 2002;13:433–438. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
