

Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases
- PMID: 16046538
- PMCID: PMC1180625
- DOI: 10.1073/pnas.0504952102
Inhibition of drug-resistant mutants of ABL, KIT, and EGF receptor kinases
Abstract
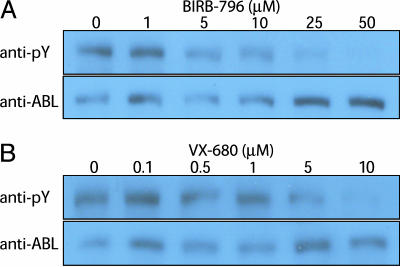
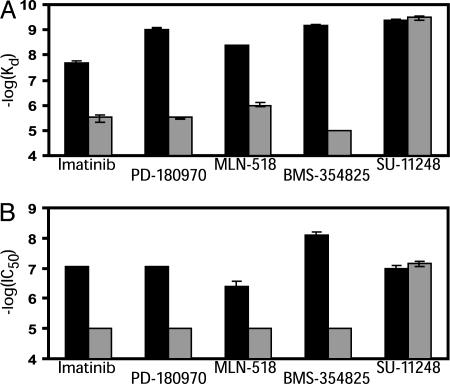
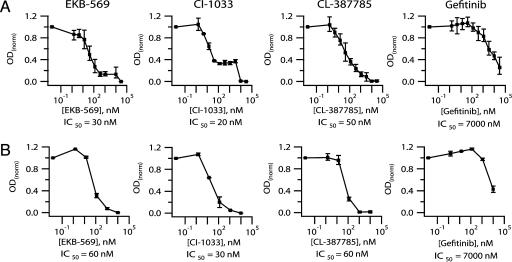
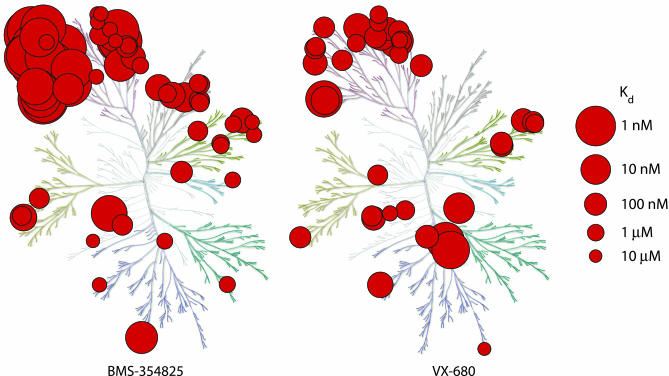
To realize the full potential of targeted protein kinase inhibitors for the treatment of cancer, it is important to address the emergence of drug resistance in treated patients. Mutant forms of BCR-ABL, KIT, and the EGF receptor (EGFR) have been found that confer resistance to the drugs imatinib, gefitinib, and erlotinib. The mutations weaken or prevent drug binding, and interestingly, one of the most common sites of mutation in all three kinases is a highly conserved "gatekeeper" threonine residue near the kinase active site. We have identified existing clinical compounds that bind and inhibit drug-resistant mutant variants of ABL, KIT, and EGFR. We found that the Aurora kinase inhibitor VX-680 and the p38 inhibitor BIRB-796 inhibit the imatinib- and BMS-354825-resistant ABL(T315I) kinase. The KIT/FLT3 inhibitor SU-11248 potently inhibits the imatinib-resistant KIT(V559D/T670I) kinase, consistent with the clinical efficacy of SU-11248 against imatinib-resistant gastrointestinal tumors, and the EGFR inhibitors EKB-569 and CI-1033, but not GW-572016 and ZD-6474, potently inhibit the gefitinib- and erlotinib-resistant EGFR(L858R/T790M) kinase. EKB-569 and CI-1033 are already in clinical trials, and our results suggest that they should be considered for testing in the treatment of gefitinib/erlotinib-resistant non-small cell lung cancer. The results highlight the strategy of screening existing clinical compounds against newly identified drug-resistant mutant variants to find compounds that may serve as starting points for the development of next-generation drugs, or that could be used directly to treat patients that have acquired resistance to first-generation targeted therapy.
Figures
References
-
- Sawyers, C. (2004) Nature 432, 294-297. - PubMed
-
- Druker, B. J. (2004) Oncologist 9, 357-360. - PubMed
-
- Wadleigh, M., DeAngelo, D. J., Griffin, J. D. & Stone, R. M. (2005) Blood 105, 22-30. - PubMed
-
- Gorre, M. E., Mohammed, M., Ellwood, K., Hsu, N., Paquette, R., Rao, P. N. & Sawyers, C. L. (2001) Science 293, 876-880. - PubMed
-
- Deininger, M., Buchdunger, E. & Druker, B. J. (2005) Blood 105, 2640-2653. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Research Materials
Miscellaneous
