

A role for the plasminogen activator system in inflammation and neurodegeneration in the central nervous system during experimental allergic encephalomyelitis
- PMID: 16049338
- PMCID: PMC1603566
- DOI: 10.1016/S0002-9440(10)62996-3
A role for the plasminogen activator system in inflammation and neurodegeneration in the central nervous system during experimental allergic encephalomyelitis
Abstract
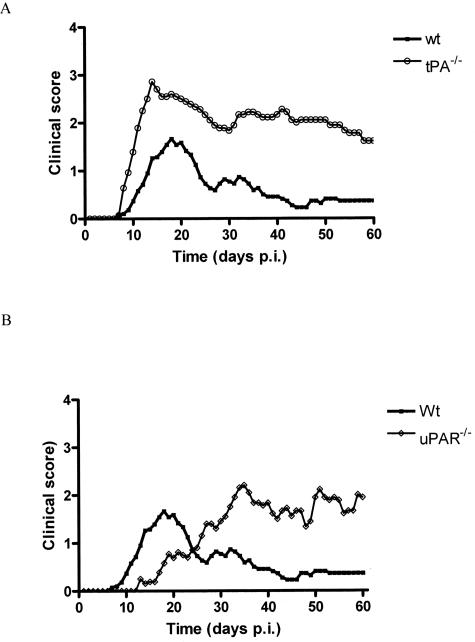
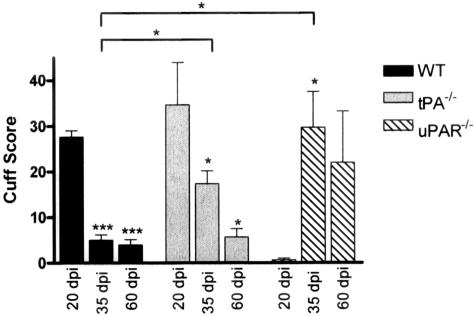
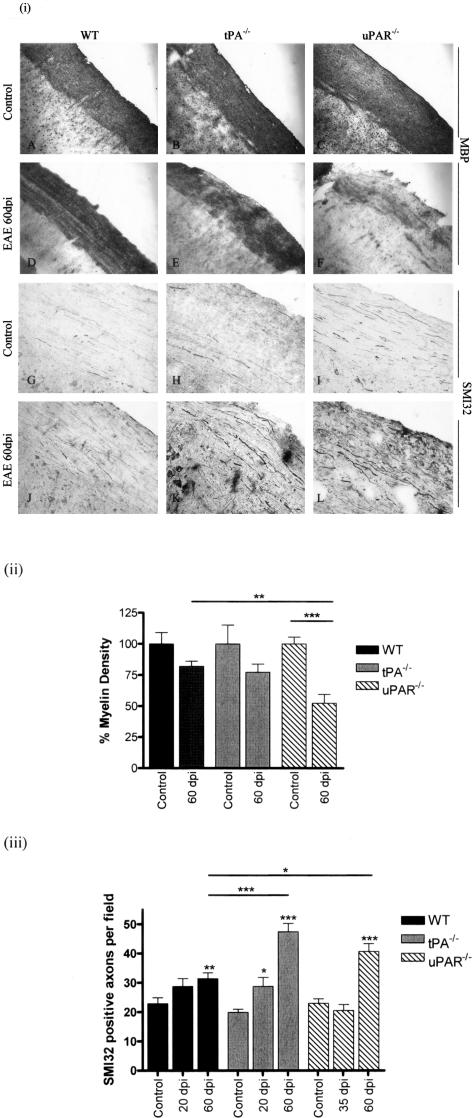
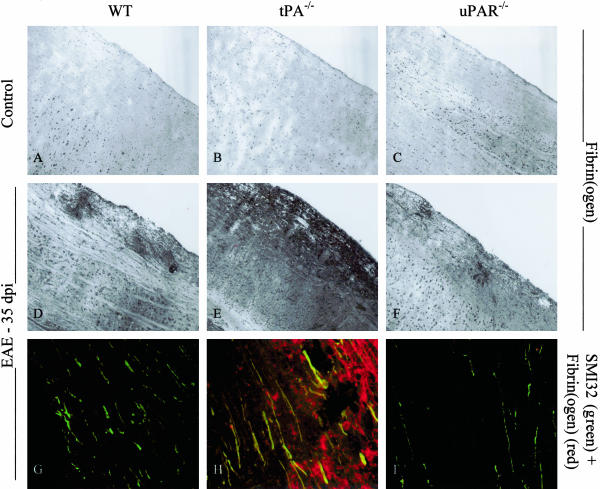
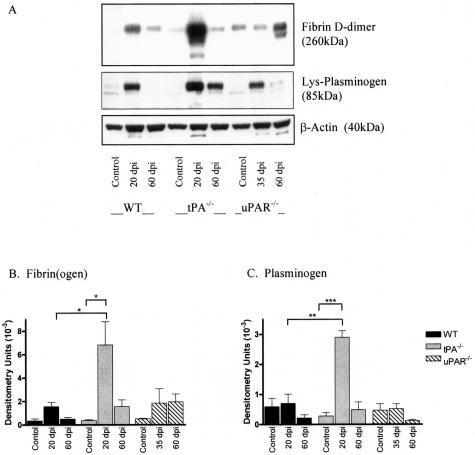
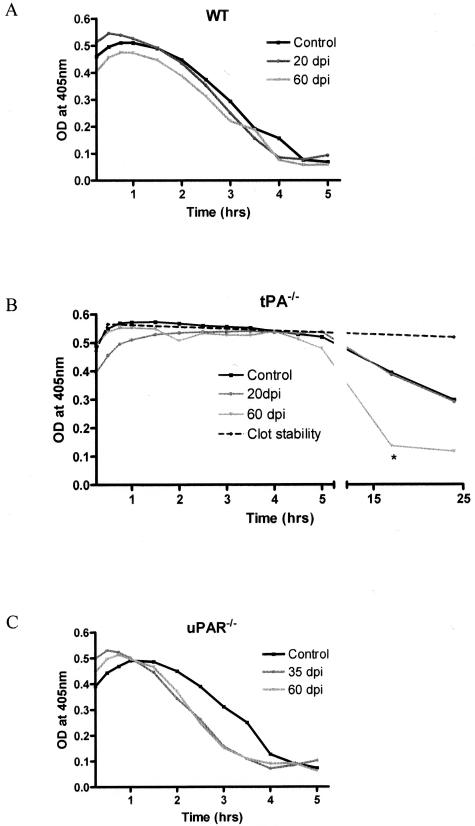
Early signs of inflammatory demyelination include entry of fibrin(ogen) into the central nervous system (CNS), which is normally excluded by the blood-brain barrier, and up-regulation of components of the plasminogen activator system. Using mice deficient in tissue-type plasminogen activator (tPA-/-) and urokinase plasminogen activator receptor (uPAR-/-), we investigated the involvement of the PA system on the clinical and pathological features of experimental allergic encephalomyelitis, an animal model of multiple sclerosis. tPA-/- mice suffered an early and a more severe acute disease characterized by incomplete recovery when compared to wild-type controls, with significantly higher CNS levels of plasminogen activator inhibitor-1. This correlated with fibrin accumulation, which co-localized with nonphosphorylated neurofilament on thickened axons in experimental allergic encephalomyelitis tissue. In contrast, uPAR-/- mice had a delayed, less acute disease reflected in delayed infiltration of inflammatory cells. These animals developed chronic disease as a result of steadily increased inflammation, increased levels of urokinase-type plasminogen activator (uPA), and greater degree of demyelination. Thus, the plasminogen activator system can modulate both inflammatory and degenerative events in the CNS through the respective effects of tPA and uPAR on fibrinolysis and cell adhesion/migration, manipulation of which may have therapeutic implications for multiple sclerosis.
Figures


Similar articles
-
Coordinated induction of extracellular proteolysis systems during experimental autoimmune encephalomyelitis in mice.Am J Pathol. 2001 Dec;159(6):2227-37. doi: 10.1016/S0002-9440(10)63073-8. Am J Pathol. 2001. PMID: 11733372 Free PMC article.
-
Chronic relapsing experimental allergic encephalomyelitis (CREAE) in plasminogen activator inhibitor-1 knockout mice: the effect of fibrinolysis during neuroinflammation.Neuropathol Appl Neurobiol. 2008 Apr;34(2):216-30. doi: 10.1111/j.1365-2990.2007.00889.x. Epub 2007 Nov 5. Neuropathol Appl Neurobiol. 2008. PMID: 17983428
-
Plasminogen activators in multiple sclerosis lesions: implications for the inflammatory response and axonal damage.Brain. 2001 Oct;124(Pt 10):1978-88. doi: 10.1093/brain/124.10.1978. Brain. 2001. PMID: 11571216
-
Regulation and interactions in the activation of cell-associated plasminogen.Cell Mol Life Sci. 2004 Nov;61(22):2840-58. doi: 10.1007/s00018-004-4230-9. Cell Mol Life Sci. 2004. PMID: 15558213 Free PMC article. Review.
-
Endothelium and disordered fibrin turnover in the injured lung: newly recognized pathways.Crit Care Med. 2002 May;30(5 Suppl):S274-80. doi: 10.1097/00003246-200205001-00017. Crit Care Med. 2002. PMID: 12004248 Review.
Cited by
-
Nonionotropic Action of Endothelial NMDA Receptors on Blood-Brain Barrier Permeability via Rho/ROCK-Mediated Phosphorylation of Myosin.J Neurosci. 2020 Feb 19;40(8):1778-1787. doi: 10.1523/JNEUROSCI.0969-19.2019. Epub 2020 Jan 17. J Neurosci. 2020. PMID: 31953371 Free PMC article.
-
The Coagulation Factors Fibrinogen, Thrombin, and Factor XII in Inflammatory Disorders-A Systematic Review.Front Immunol. 2018 Jul 26;9:1731. doi: 10.3389/fimmu.2018.01731. eCollection 2018. Front Immunol. 2018. PMID: 30105021 Free PMC article.
-
Roles of the tissue-type plasminogen activator in immune response.Cell Immunol. 2022 Jan;371:104451. doi: 10.1016/j.cellimm.2021.104451. Epub 2021 Nov 6. Cell Immunol. 2022. PMID: 34781155 Free PMC article. Review.
-
A critical role for plasminogen in inflammation.J Exp Med. 2020 Apr 6;217(4):e20191865. doi: 10.1084/jem.20191865. J Exp Med. 2020. PMID: 32159743 Free PMC article. Review.
-
Plasminogen Deficiency Delays the Onset and Protects from Demyelination and Paralysis in Autoimmune Neuroinflammatory Disease.J Neurosci. 2017 Apr 5;37(14):3776-3788. doi: 10.1523/JNEUROSCI.2932-15.2017. Epub 2017 Mar 8. J Neurosci. 2017. PMID: 28275164 Free PMC article.
References
-
- Lo EH, Wang X, Cuzner ML. Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res. 2002;69:1–9. - PubMed
-
- Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J. The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol. 1996;55:1194–1204. - PubMed
-
- Kermode AG, Thompson AJ, Tofts P, MacManus DG, Kendall BE, Kingsley DP, Moseley IF, Rudge P, McDonald WI. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenic and clinical implications. Brain. 1990;113:1477–1489. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
