

Caveola-dependent endocytic entry of amphotropic murine leukemia virus
- PMID: 16051869
- PMCID: PMC1182675
- DOI: 10.1128/JVI.79.16.10776-10787.2005
Caveola-dependent endocytic entry of amphotropic murine leukemia virus
Abstract
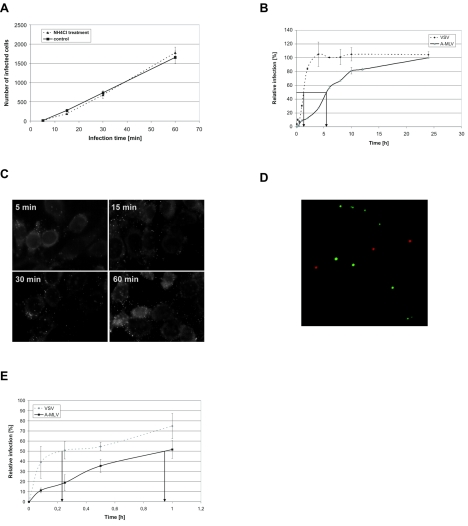
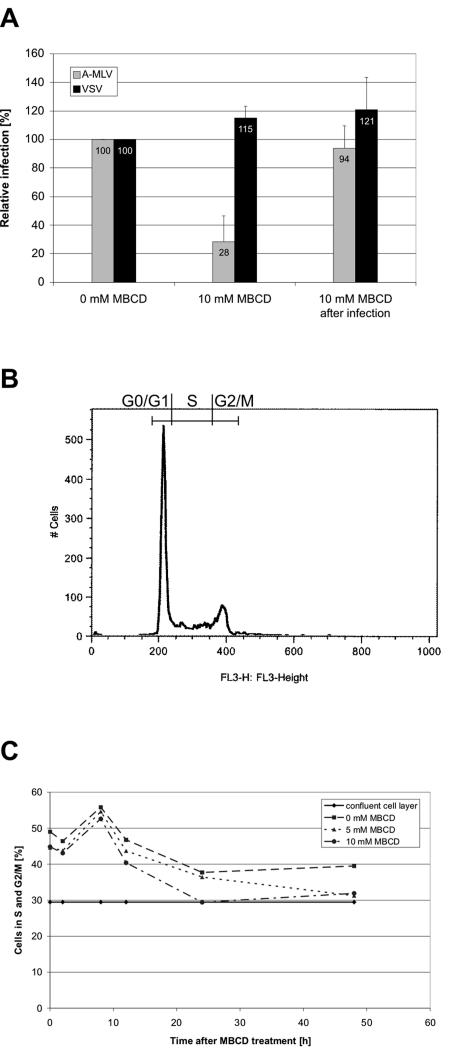
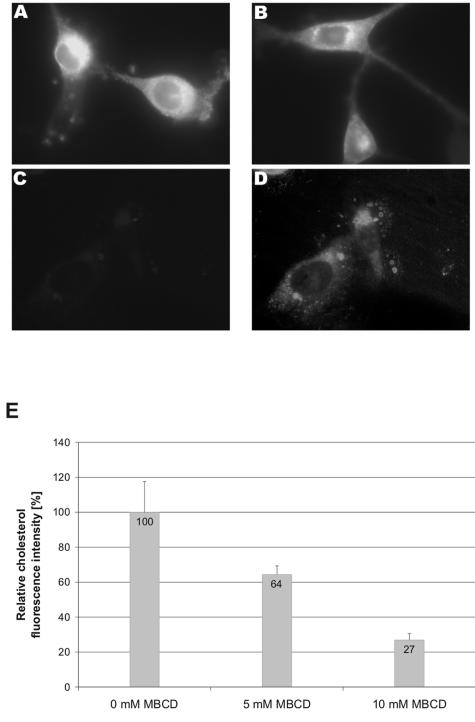
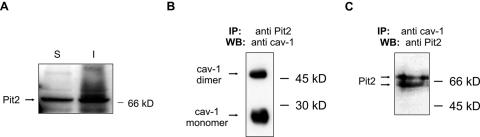
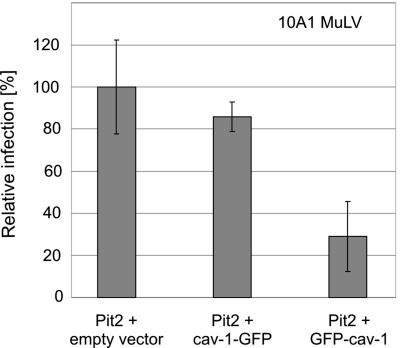
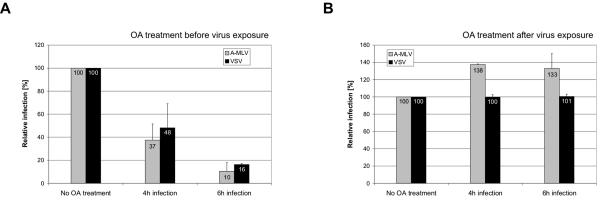
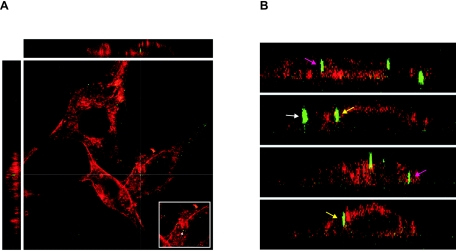
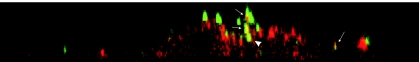
Early results suggested that the amphotropic murine leukemia virus (A-MLV) does not enter cells via endocytosis through clathrin-coated pits and this gammaretrovirus has therefore been anticipated to fuse directly with the plasma membrane. However, here we present data implicating a caveola-mediated endocytic entry route for A-MLV via its receptor Pit2. Caveolae belong to the cholesterol-rich microdomains characterized by resistance to nonionic detergents such as Triton X-100. Extraction of murine fibroblastic NIH 3T3 cells in cold Triton X-100 showed the presence of the A-MLV receptor Pit2 in detergent-insoluble microdomains. Using coimmunoprecipitation of cell extracts, we were able to demonstrate direct association of Pit2 with caveolin-1, the structural protein of caveolae. Other investigations revealed that A-MLV infection in contrast to vesicular stomatitis virus infection is a slow process (t(1/2) approximately 5 h), which is dependent on plasma membrane cholesterol but independent of NH4Cl treatment of cells; NH4Cl impairs entry via clathrin-coated pits. Furthermore, expression of dominant-negative caveolin-1 decreased the susceptibility to infection via Pit2 by approximately 70%. These results show that A-MLV can enter cells via a caveola-dependent entry route. Moreover, increase in A-MLV infection by treatment with okadaic acid as well as entry of fusion-defective fluorescent A-MLV virions in NIH 3T3 cells further confirmed our findings and show that A-MLV can enter mouse fibroblasts via an endocytic entry route involving caveolae. Finally, we also found colocalization of fusion-defective fluorescent A-MLV virions with caveolin-1 in NIH 3T3 cells. This is the first time substantial evidence has been presented implicating the existence of a caveola-dependent endocytic entry pathway for a retrovirus.
Figures
References
-
- Anderson, R. G. W. 1998. The caveolae membrane system. Annu. Rev. Biochem. 67:199-225. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
