

Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin
- PMID: 16080118
- PMCID: PMC1226208
- DOI: 10.1086/444548
Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin
Abstract
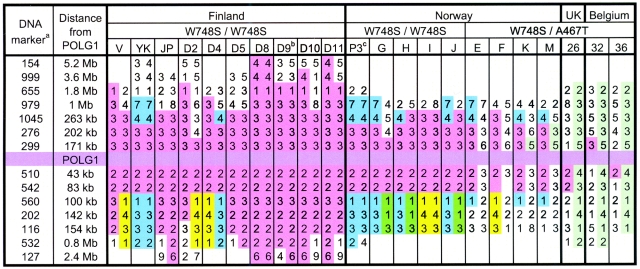
Mutations in the catalytic subunit of the mitochondrial DNA polymerase gamma (POLG) have been found to be an important cause of neurological disease. Recently, we and collaborators reported a new neurodegenerative disorder with autosomal recessive ataxia in four patients homozygous for two amino acid changes in POLG: W748S in cis with E1143G. Here, we studied the frequency of this allele and found it to be among the most common genetic causes of inherited ataxia in Finland. We identified 27 patients with mitochondrial recessive ataxia syndrome (MIRAS) from 15 Finnish families, with a carrier frequency in the general population of 1 : 125. Since the mutation pair W748S+E1143G has also been described in European patients, we examined the haplotypes of 13 non-Finnish, European patients with the W748S mutation. Haplotype analysis revealed that all the chromosomes carrying these two changes, in patients from Finland, Norway, the United Kingdom, and Belgium, originate from a common ancient founder. In Finland and Norway, long, common, northern haplotypes, outside the core haplotype, could be identified. Despite having identical homozygous mutations, the Finnish patients with this adult- or juvenile-onset disease had surprisingly heterogeneous phenotypes, albeit with a characteristic set of features, including ataxia, peripheral neuropathy, dysarthria, mild cognitive impairment, involuntary movements, psychiatric symptoms, and epileptic seizures. The high carrier frequency in Finland, the high number of patients in Norway, and the ancient European founder chromosome indicate that this newly identified ataxia should be considered in the first-line differential diagnosis of progressive ataxia syndromes.
Figures

References
Web Resources
-
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/ (for SNPs rs2307438, rs2302084, rs2246900, rs2307449, rs2307433, rs2072266, rs2072267, and rs3176183)
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for POLG1 [accession number NM_002693]; sequence numbering starts from ATG)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for POLG1; PEO; SCA1, 2, 3, 6, 7, 8, 10, 12, and 17; DRPLA; and FRDA)
References
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, et al (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427 - PubMed
-
- Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335:1169–1175 - PubMed
-
- Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, Carrara F, Parini R, Simonati A, Santer R, Zeviani M (2005) Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-γA. Brain 128:723–731 - PubMed
-
- Foury F, Cazzalini O (1997) Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett 411:373–377 - PubMed
-
- Harding AE (1981) Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104:589–620 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
