

Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males
- PMID: 16080119
- PMCID: PMC1226209
- DOI: 10.1086/444549
Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males
Abstract
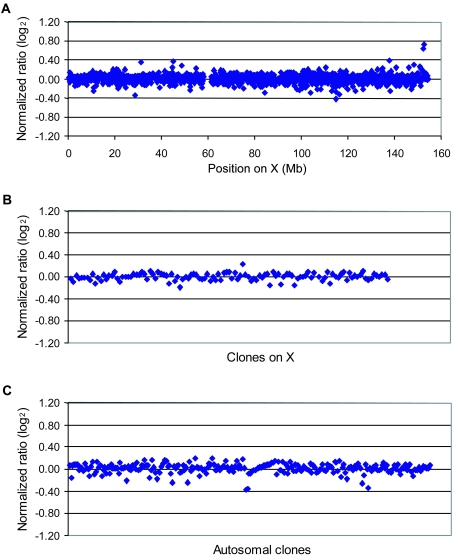
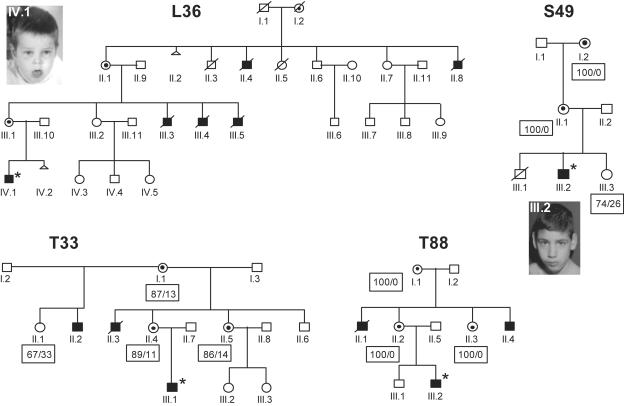
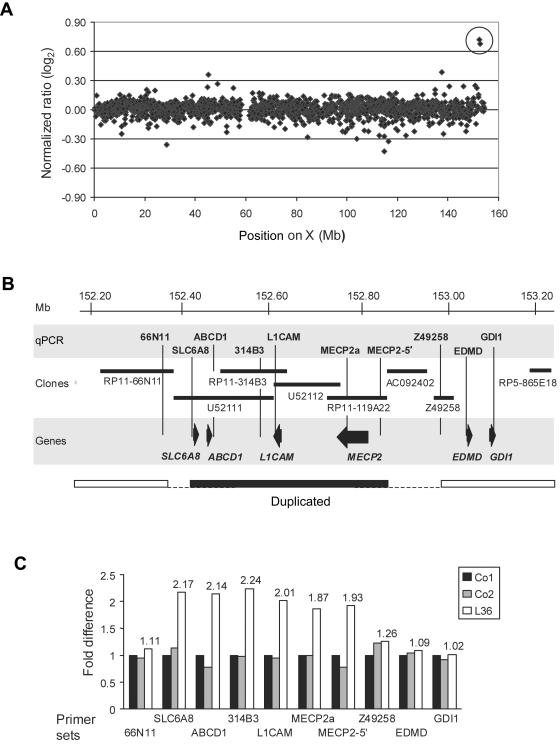
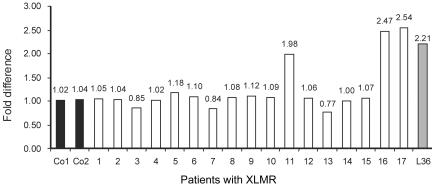
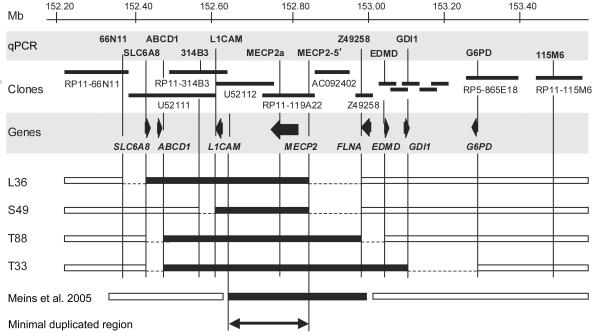
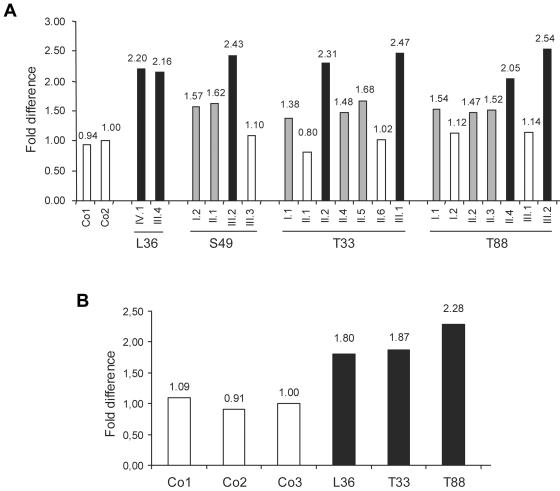
Loss-of-function mutations of the MECP2 gene at Xq28 are associated with Rett syndrome in females and with syndromic and nonsyndromic forms of mental retardation (MR) in males. By array comparative genomic hybridization (array-CGH), we identified a small duplication at Xq28 in a large family with a severe form of MR associated with progressive spasticity. Screening by real-time quantitation of 17 additional patients with MR who have similar phenotypes revealed three more duplications. The duplications in the four patients vary in size from 0.4 to 0.8 Mb and harbor several genes, which, for each duplication, include the MR-related L1CAM and MECP2 genes. The proximal breakpoints are located within a 250-kb region centromeric of L1CAM, whereas the distal breakpoints are located in a 300-kb interval telomeric of MECP2. The precise size and location of each duplication is different in the four patients. The duplications segregate with the disease in the families, and asymptomatic carrier females show complete skewing of X inactivation. Comparison of the clinical features in these patients and in a previously reported patient enables refinement of the genotype-phenotype correlation and strongly suggests that increased dosage of MECP2 results in the MR phenotype. Our findings demonstrate that, in humans, not only impaired or abolished gene function but also increased MeCP2 dosage causes a distinct phenotype. Moreover, duplication of the MECP2 region occurs frequently in male patients with a severe form of MR, which justifies quantitative screening of MECP2 in this group of patients.
Figures
References
Web Resources
-
- Ensembl Genome Browser, http://www.ensembl.org/Homo_sapiens/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MECP2 and L1CAM and Rett, HSAS, MASA, and CRASH syndromes)
-
- UCSC Human Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway?org=Human
References
-
- Akiyama M, Kawame H, Ohashi H, Tohma T, Ohta H, Shishikura A, Miyata I, Usui N, Eto Y (2001) Functional disomy for Xq26.3-qter in a boy with an unbalanced t(X;21)(q26.3;p11.2) translocation. Am J Med Genet 99:111–114 - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188 - PubMed
-
- Amir RE, Zoghbi HY (2000) Rett syndrome: methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am J Med Genet 97:147–152 - PubMed
-
- Ariani F, Mari F, Pescucci C, Longo I, Bruttini M, Meloni I, Hayek G, Rocchi R, Zappella M, Renieri A (2004) Real-time quantitative PCR as a routine method for screening large rearrangements in Rett syndrome: report of one case of MECP2 deletion and one case of MECP2 duplication. Hum Mutat 24:172–177 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
