

Correcting common errors in identifying cancer-specific serum peptide signatures
- PMID: 16083255
- PMCID: PMC1852495
- DOI: 10.1021/pr050034b
Correcting common errors in identifying cancer-specific serum peptide signatures
Abstract
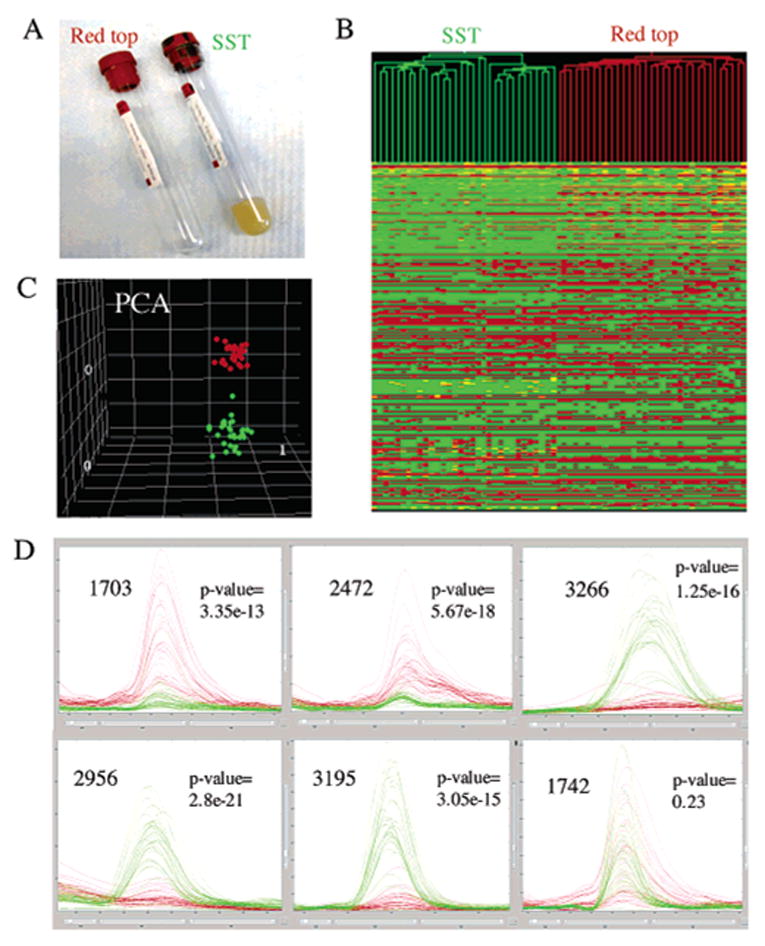
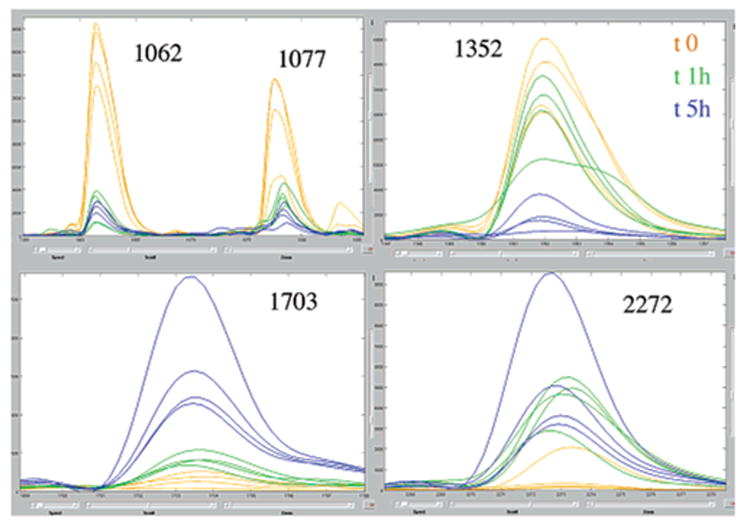
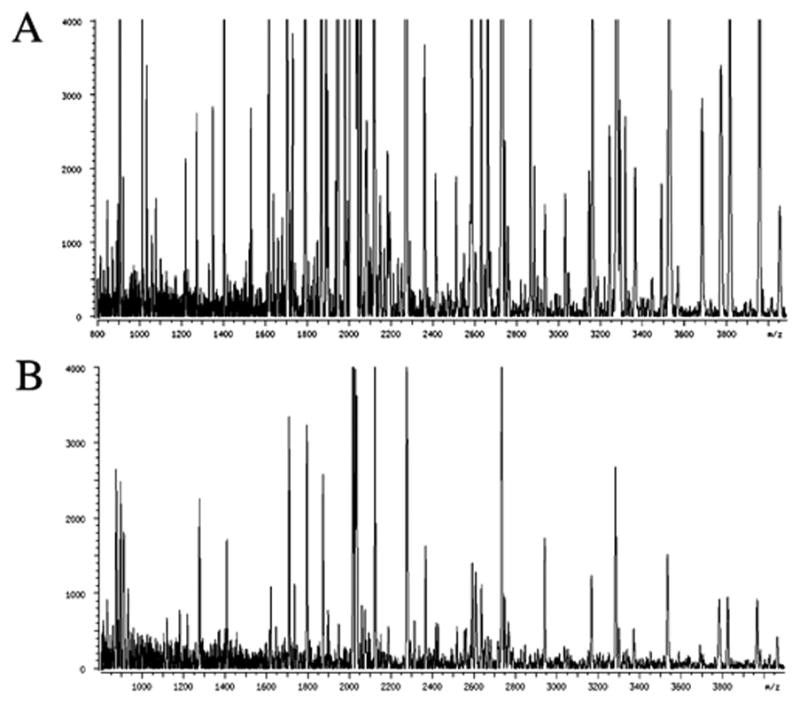
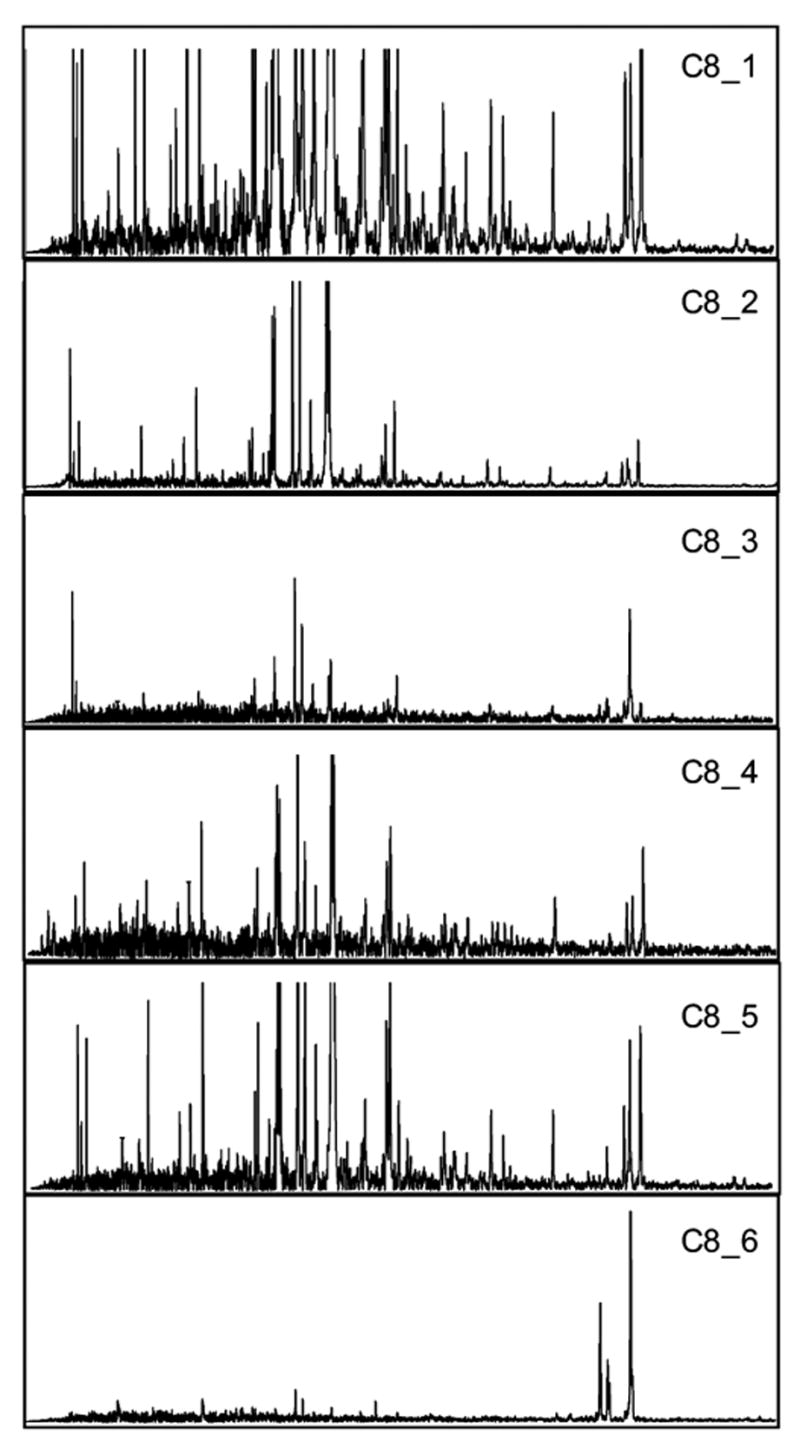
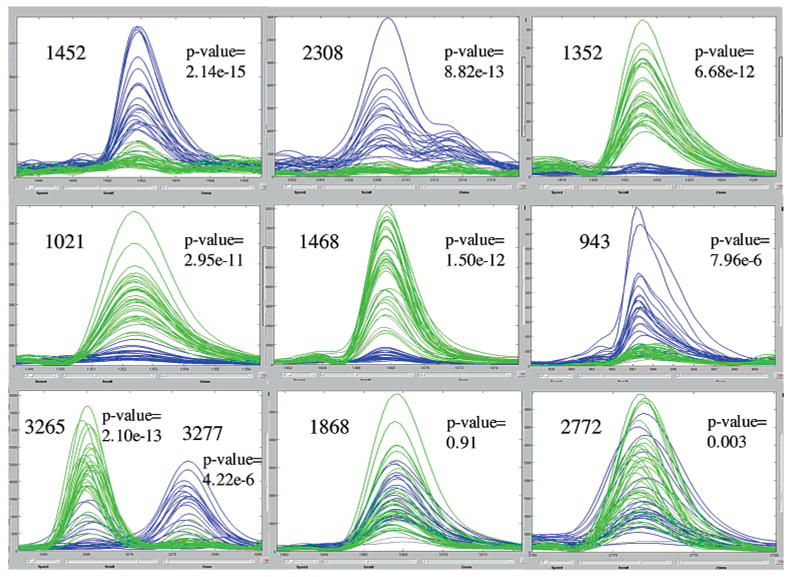
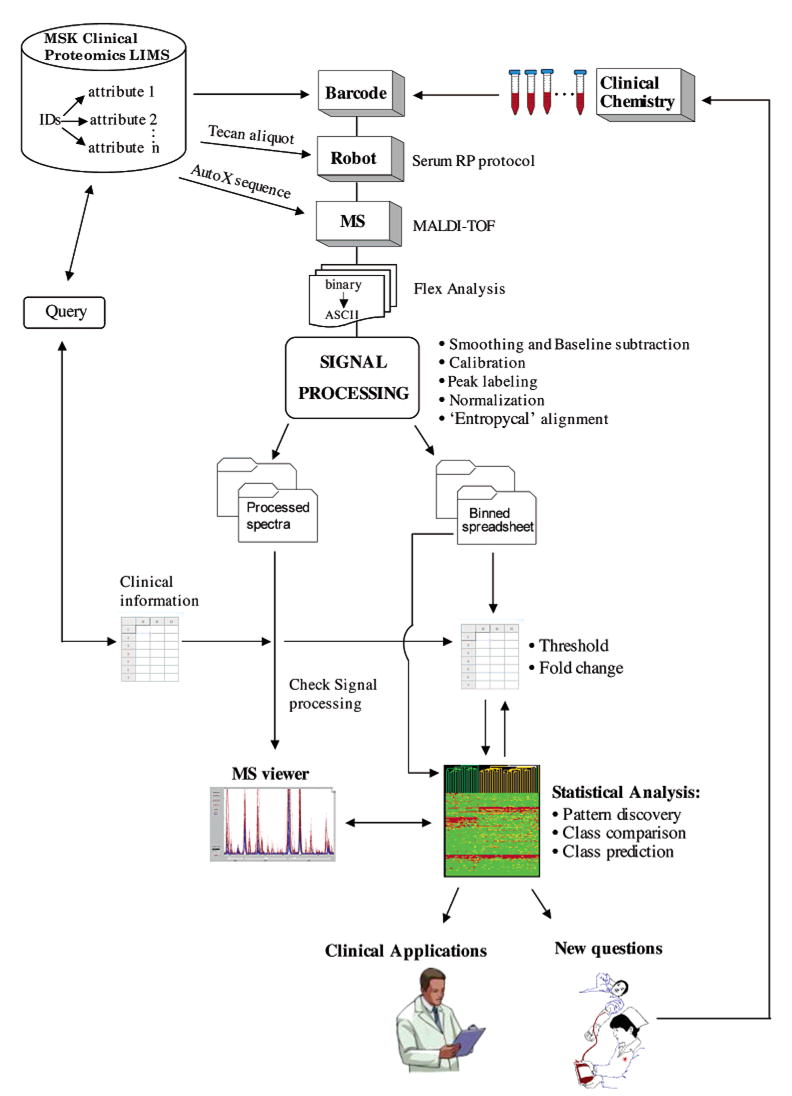
"Molecular signatures" are the qualitative and quantitative patterns of groups of biomolecules (e.g., mRNA, proteins, peptides, or metabolites) in a cell, tissue, biological fluid, or an entire organism. To apply this concept to biomarker discovery, the measurements should ideally be noninvasive and performed in a single read-out. We have therefore developed a peptidomics platform that couples magnetics-based, automated solid-phase extraction of small peptides with a high-resolution MALDI-TOF mass spectrometric readout (Villanueva, J.; Philip, J.; Entenberg, D.; Chaparro, C. A.; Tanwar, M. K.; Holland, E. C.; Tempst, P. Anal. Chem. 2004, 76, 1560-1570). Since hundreds of peptides can be detected in microliter volumes of serum, it allows to search for disease signatures, for instance in the presence of cancer. We have now evaluated, optimized, and standardized a number of clinical and analytical chemistry variables that are major sources of bias; ranging from blood collection and clotting, to serum storage and handling, automated peptide extraction, crystallization, spectral acquisition, and signal processing. In addition, proper alignment of spectra and user-friendly visualization tools are essential for meaningful, certifiable data mining. We introduce a minimal entropy algorithm, "Entropycal", that simplifies alignment and subsequent statistical analysis and increases the percentage of the highly distinguishing spectral information being retained after feature selection of the datasets. Using the improved analytical platform and tools, and a commercial statistics program, we found that sera from thyroid cancer patients can be distinguished from healthy controls based on an array of 98 discriminant peptides. With adequate technological and computational methods in place, and using rigorously standardized conditions, potential sources of patient related bias (e.g., gender, age, genetics, environmental, dietary, and other factors) may now be addressed.
Figures
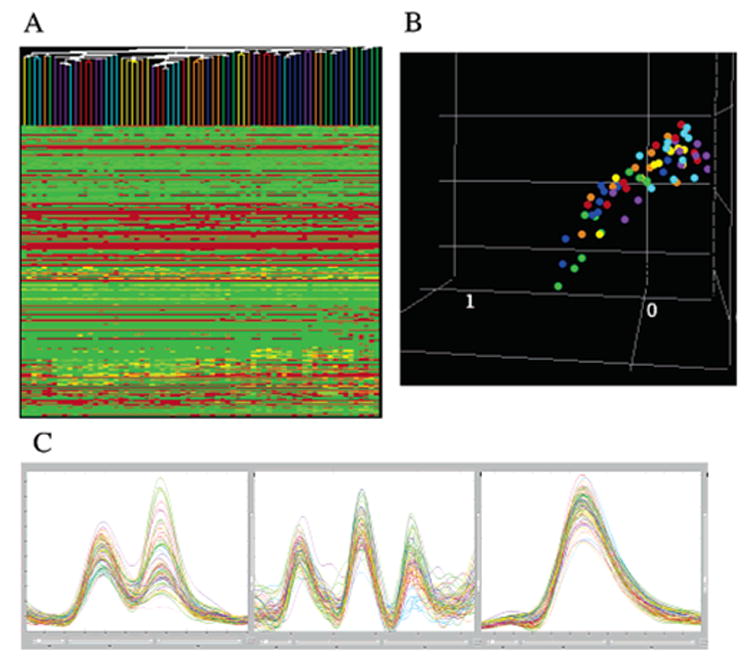
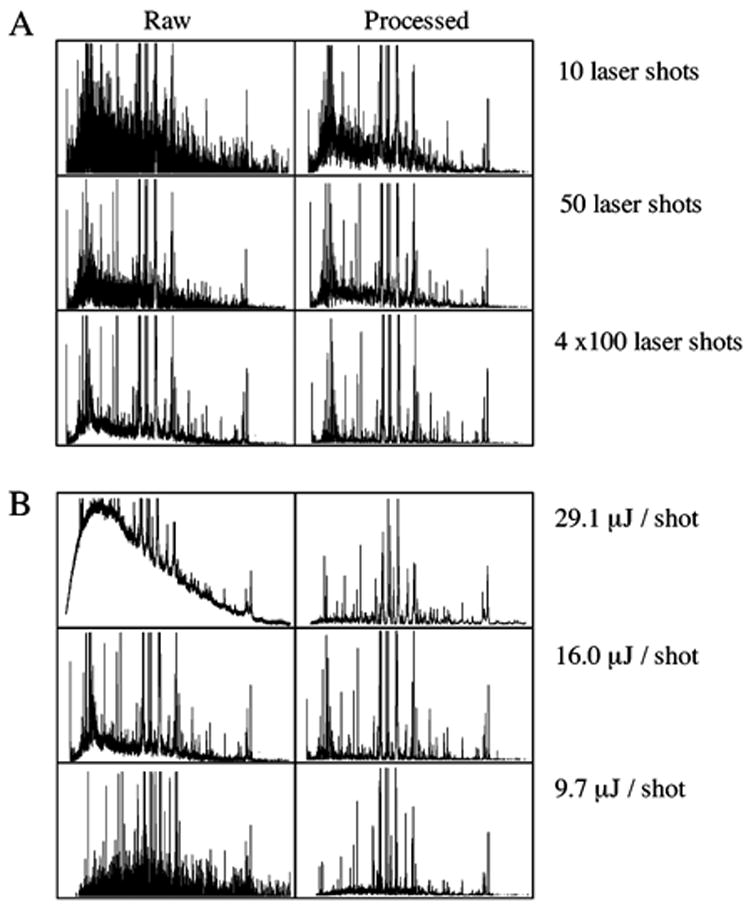
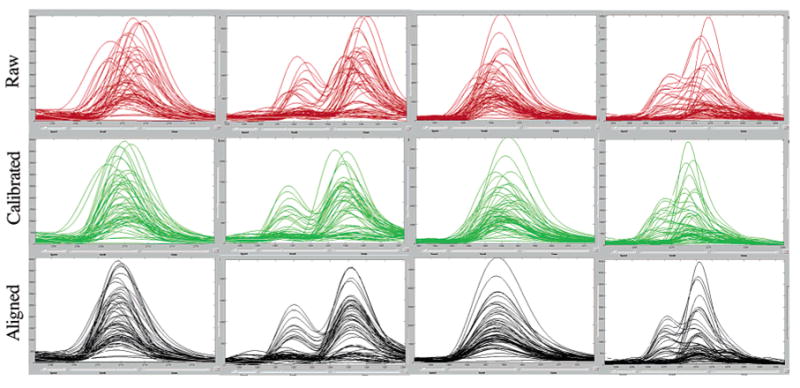
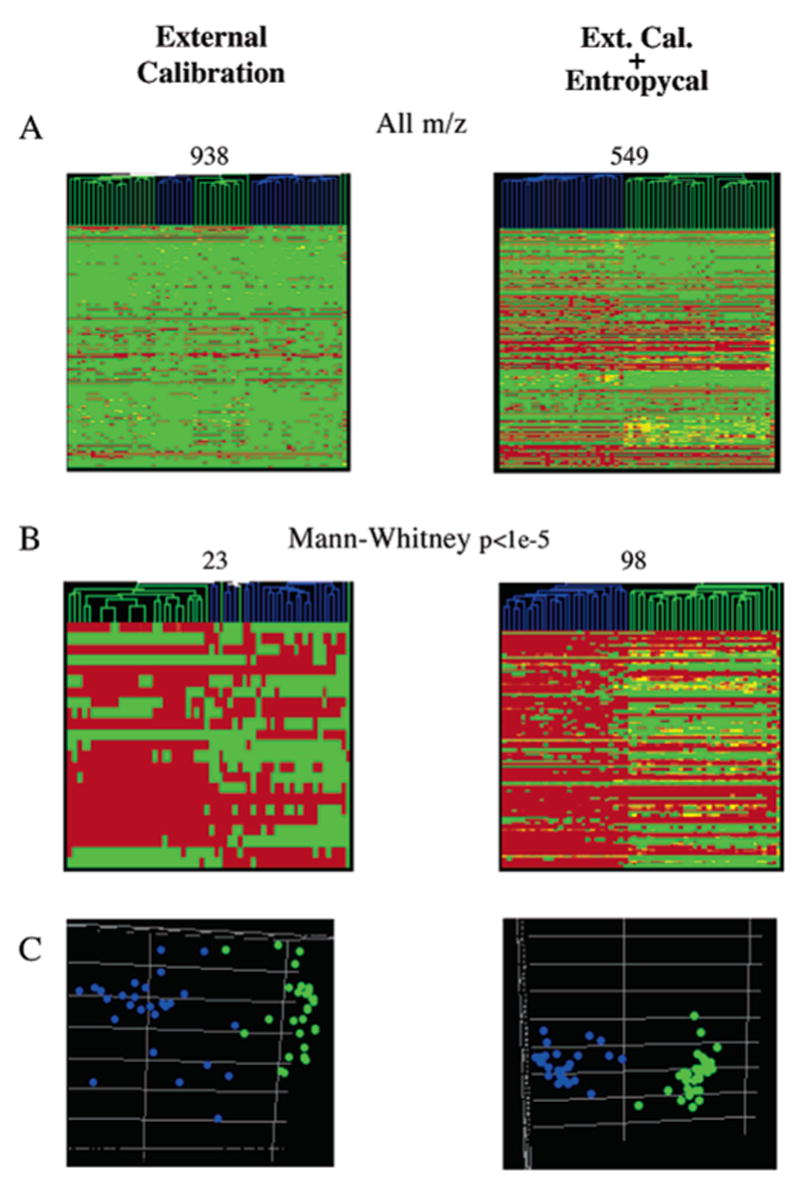
References
-
- Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, Mills GB, Simone C, Fishman DA, Kohn EC, Liotta LA. Lancet. 2002;359(9306):572–577. - PubMed
-
- Adam BL, Qu Y, Davis JW, Ward MD, Clements MA, Cazares LH, Semmes OJ, Schellhammer PF, Yasui Y, Feng Z, Wright GL., Jr Cancer Res. 2002;62(13):3609–3614. - PubMed
-
- Li J, Zhang Z, Rosenzweig J, Wang YY, Chan DW. Clin Chem. 2002;48(8):1296–1304. - PubMed
-
- Borozdenkova S, Westbrook JA, Patel V, Wait R, Bolad I, Burke MM, Bell AD, Banner NR, Dunn MJ, Rose ML. J Proteome Res. 2004;3(2):282–288. - PubMed
-
- Ebert MP, Meuer J, Wiemer JC, Schulz HU, Reymond MA, Traugott U, Malfertheiner P, Rocken C. J Proteome Res. 2004;3(6):1261–1266. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
