

Predicting specificity-determining residues in two large eukaryotic transcription factor families
- PMID: 16085755
- PMCID: PMC1183107
- DOI: 10.1093/nar/gki755
Predicting specificity-determining residues in two large eukaryotic transcription factor families
Abstract
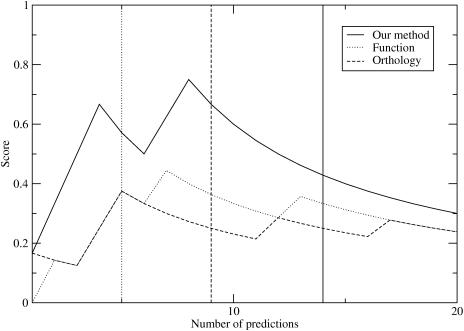
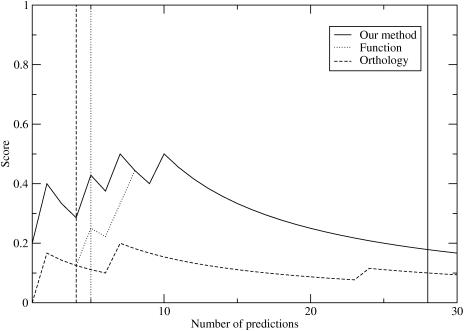
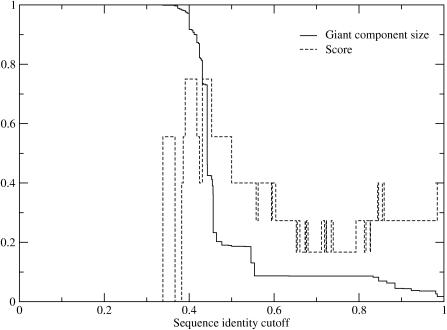
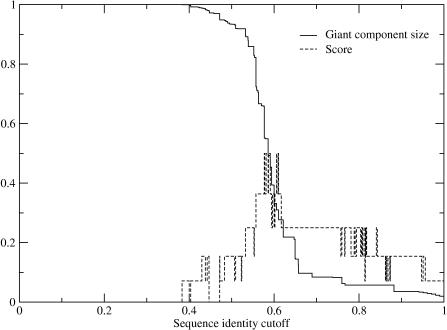
Certain amino acid residues in a protein, when mutated, change the protein's function. We present an improved method of finding these specificity-determining positions that uses all the protein sequence data available for a family of homologous proteins. We study in detail two families of eukaryotic transcription factors, basic leucine zippers and nuclear receptors, because of the large amount of sequences and experimental data available. These protein families also have a clear definition of functional specificity: DNA-binding specificity. We compare our results to three other methods, including the evolutionary trace algorithm and a method that depends on orthology relationships. All of the predictions are compared to the available mutational and crystallographic data. We find that our method provides superior predictions of the known specificity-determining residues and also predicts residue positions within these families that deserve further study for their roles in functional specificity.
Figures
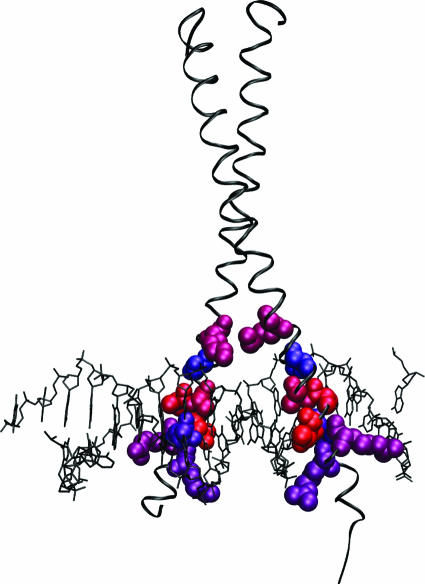
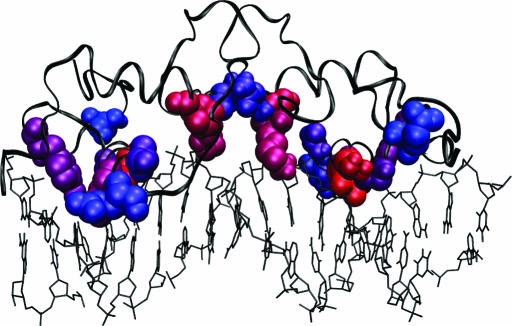
References
-
- Lichtarge O., Bourne H.R., Cohen F.E. An evolutionary trace method defines binding surfaces common to protein families. J. Mol. Biol. 1996;257:342–358. - PubMed
-
- Lichtarge O., Yamamoto K.R., Cohen F.E. Identification of functional surfaces of the zinc binding domains of intracellular receptors. J. Mol. Biol. 1997;274:325–337. - PubMed
-
- Mirny L.A., Gelfand M.S. Using orthologous and paralogous proteins to identify specificity-determining residues in bacterial transcription factors. J. Mol. Biol. 2002;321:7–20. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
