

Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis
- PMID: 16085863
- PMCID: PMC1183343
- DOI: 10.1128/AEM.71.8.4679-4689.2005
Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis
Abstract
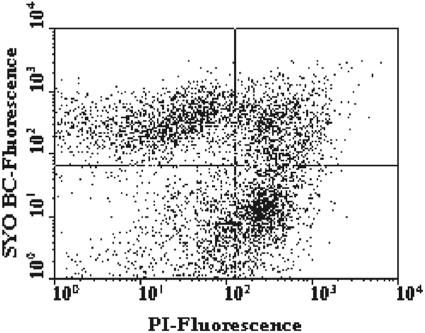
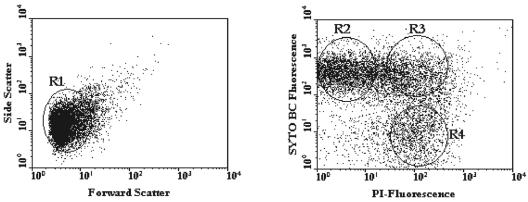
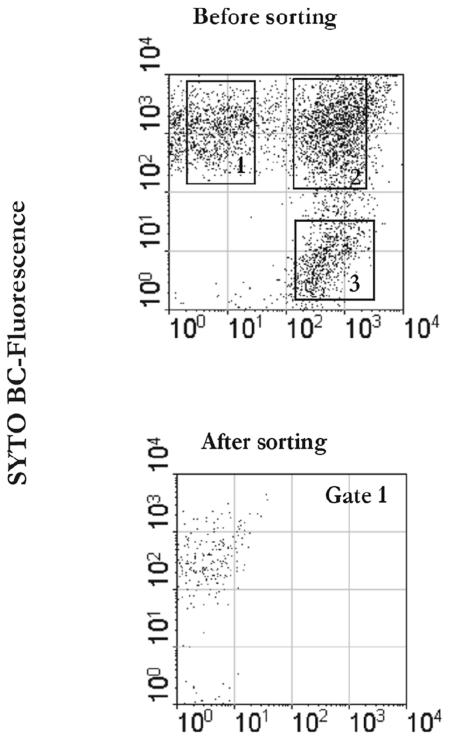
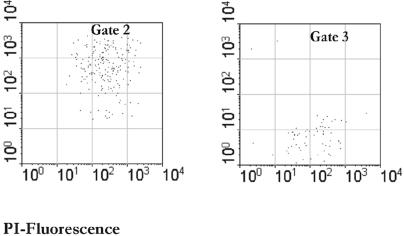
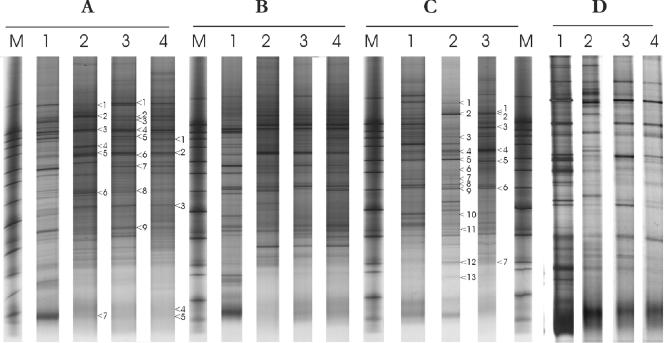
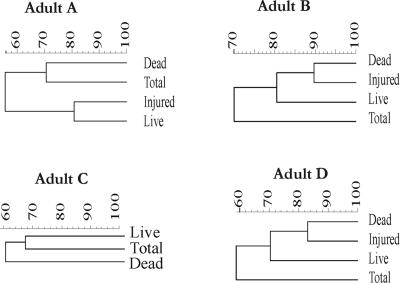
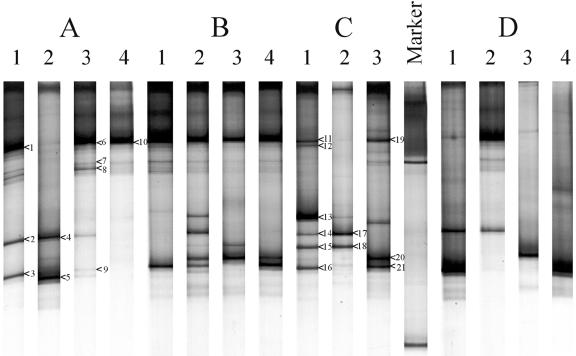
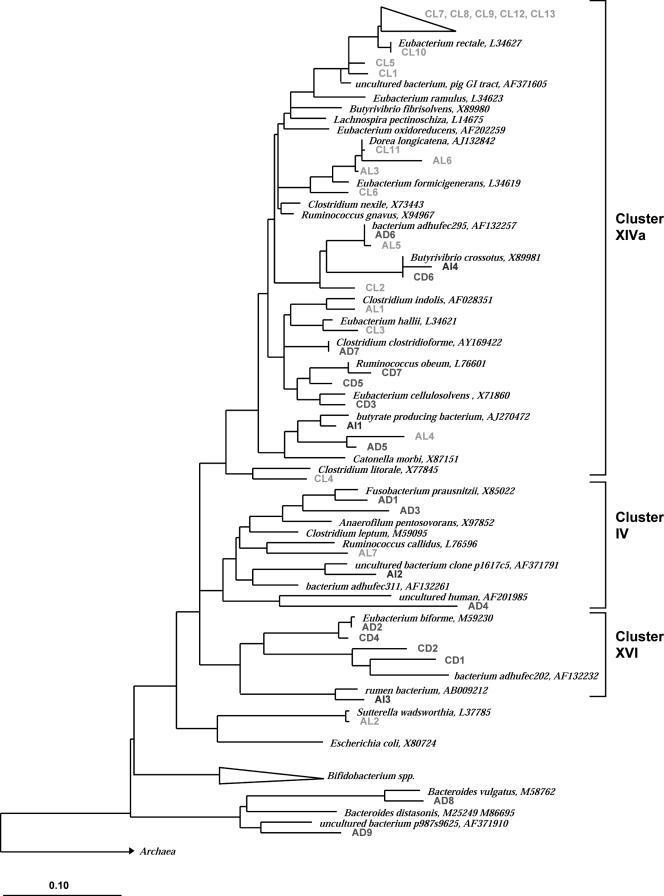
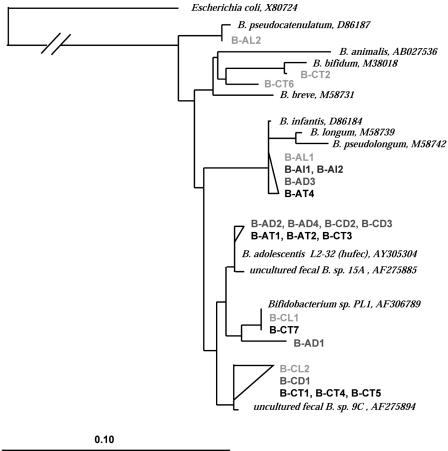
A novel approach combining a flow cytometric in situ viability assay with 16S rRNA gene analysis was used to study the relationship between diversity and activity of the fecal microbiota. Simultaneous staining with propidium iodide (PI) and SYTO BC provided clear discrimination between intact cells (49%), injured or damaged cells (19%), and dead cells (32%). The three subpopulations were sorted and characterized by denaturing gradient gel electrophoresis (DGGE) of 16S rRNA gene amplicons obtained from the total and bifidobacterial communities. This analysis revealed that not only the total community but also the distinct subpopulations are characteristic for each individual. Cloning and sequencing of the dominant bands of the DGGE patterns showed that most of clones retrieved from the live, injured, and dead fractions belonged to Clostridium coccoides, Clostridium leptum, and Bacteroides. We found that some of the butyrate-producing related bacteria, such as Eubacterium rectale and Eubacterium hallii, were obviously viable at the time of sampling. However, amplicons affiliated with Bacteroides and Ruminococcus obeum- and Eubacterium biforme-like bacteria, as well as Butyrivibrio crossotus, were obtained especially from the dead population. Furthermore, some bacterial clones were recovered from all sorted fractions, and this was especially noticeable for the Clostridium leptum cluster. The bifidobacterial phylotypes identified in total samples and sorted fractions were assigned to Bifidobacterium adolescentis, Bifidobacterium longum, Bifidobacterium infantis, Bifidobacterium pseudocatenulatum, and Bifidobacterium bifidum. Phylogenetic analysis of the live, dead, and injured cells revealed a remarkable physiological heterogeneity within these bacterial populations; B. longum and B. infantis were retrieved from all sorted fractions, while B. adolescentis was recovered mostly from the sorted dead fraction.
Figures
References
-
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. - PubMed
-
- Bäckhed, F., R. E. Ley, J. L. Sonnenburg, D. A. Peterson, and J. I. Gordon. 2005. Host-bacterial mutualism in the human intestine. Science 307:1915-1920. - PubMed
-
- Barbesti, S., S. Citterio, M. Labra, M. D. Baroni, M. G. Neri, and S. Sgorbati. 2000. Two- and three-color fluorescence flow cytometric analysis of immunoidentified viable bacteria. Cytometry 40:214-218. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
