

The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors
- PMID: 16091975
- PMCID: PMC12161179
- DOI: 10.1007/s00432-005-0002-7
The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors
Abstract
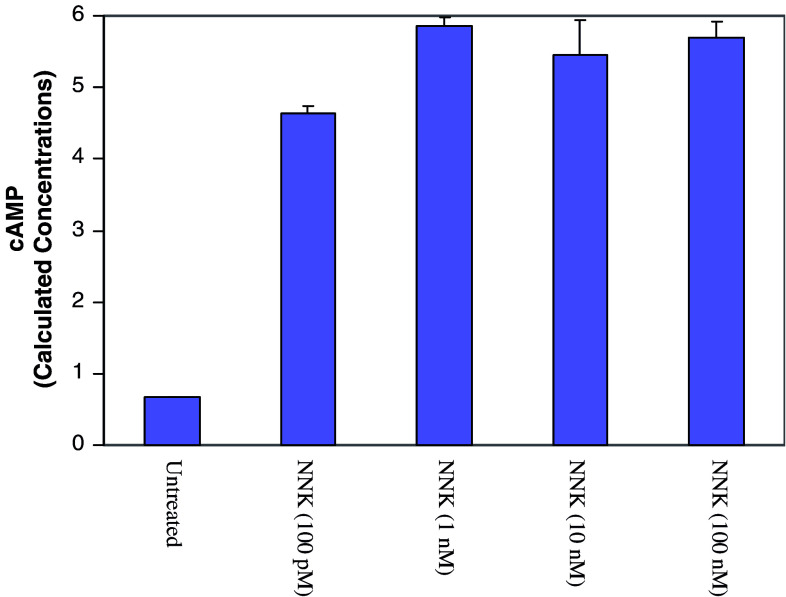
Purpose: Pancreatic ductal adenocarcinoma is an aggressive smoking-associated human cancer in both men and women. The nicotine-derived 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is thought to contribute to the development of these neoplasms in smokers through genotoxic effects. However, NNK has been recently identified as an agonist for both beta(1)- and beta(2)-adrenergic receptors. Binding of NNK to these receptors stimulates proliferation of pulmonary and pancreatic adenocarcinomas cells in vitro and in hamster models. The goal of this study was to elucidate the NNK effects on the signal transduction pathways downstream of both beta(1)- and beta(2)-adrenergic receptors in immortalized human pancreatic HPDE6-c7 cells.
Methods: The HPDE6-c7 cells are developed from normal pancreatic duct epithelial cells which are the putative cells of origin of pancreatic ductal adenocarcinoma. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazoliumbromide (MTT) cell proliferation assays, Western blot and cyclic AMP assays were employed to demonstrate the effects of NNK and other beta(1)- and beta(2)-adrenergic agonists and antagonist treatments on these cells.
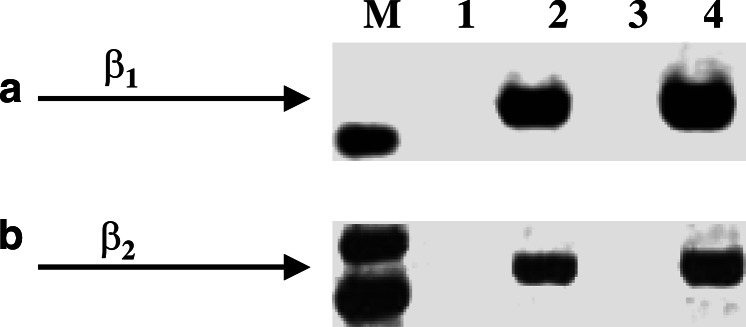
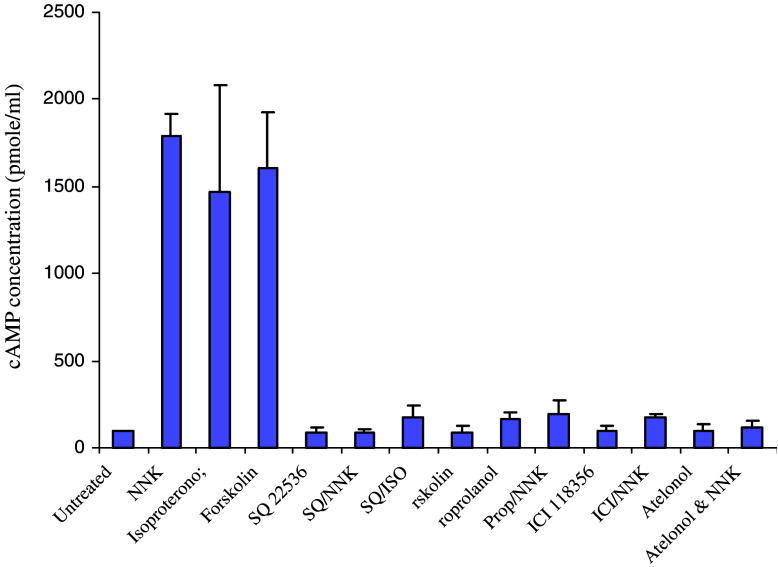
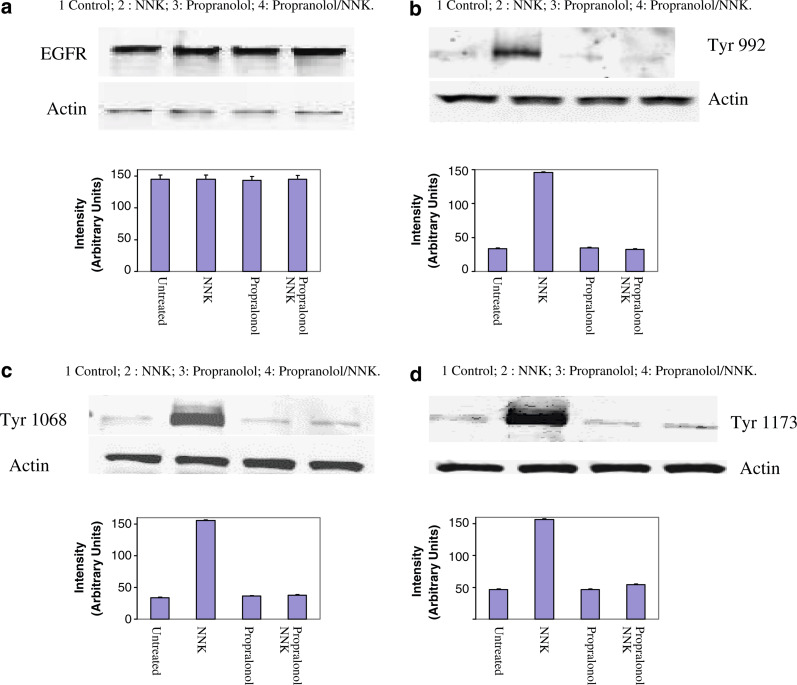
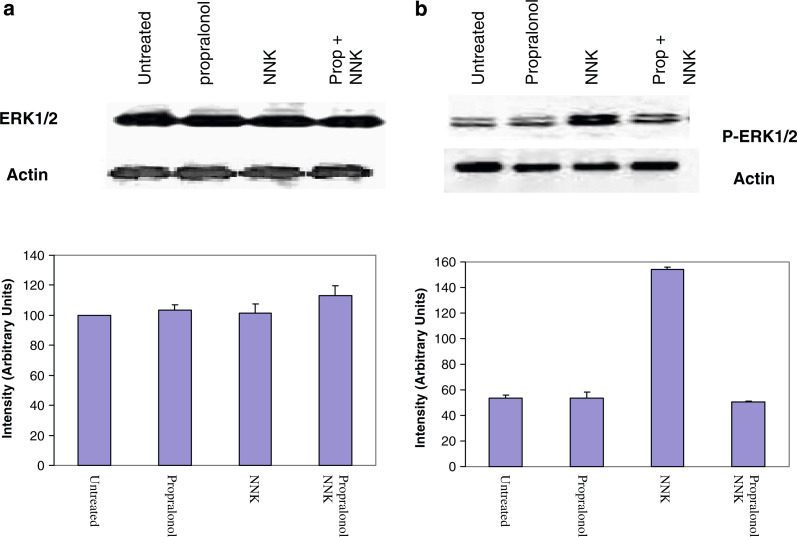
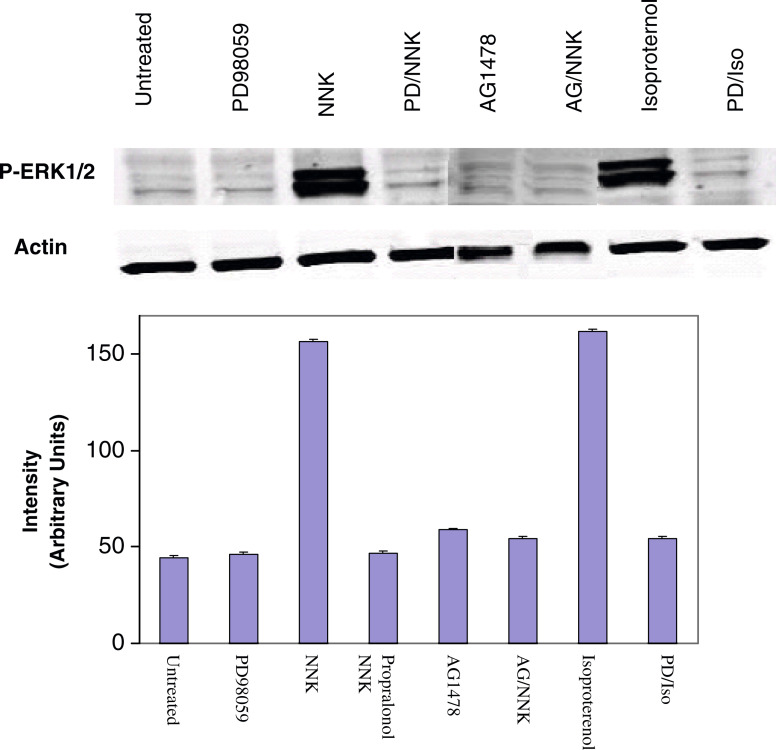
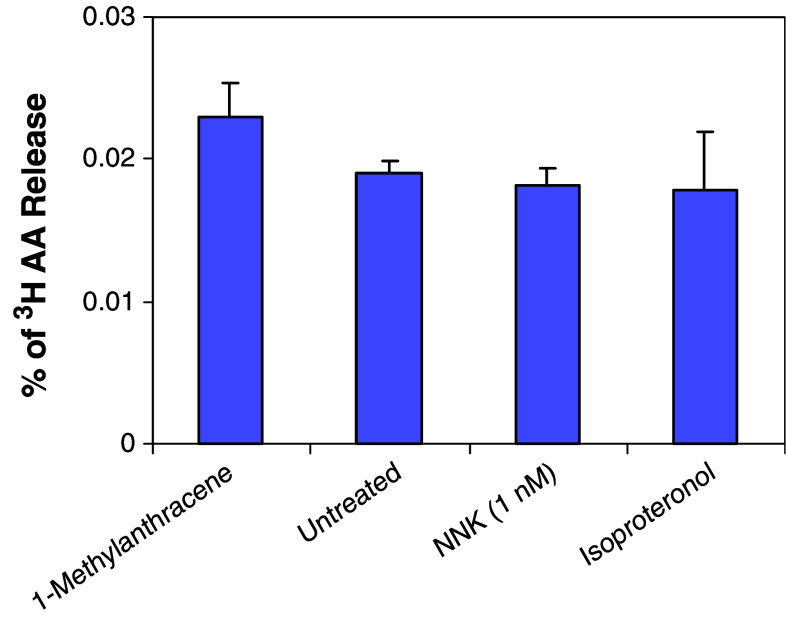
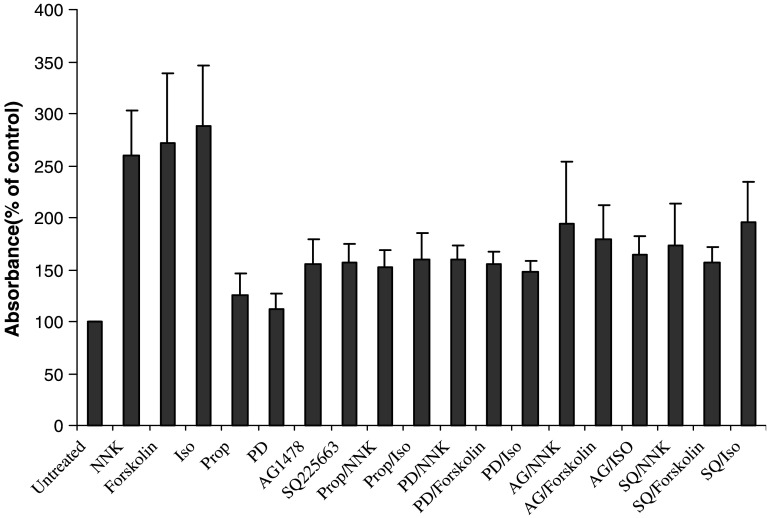
Results: MTT cell proliferation assays demonstrated that NNK and the classic beta-adrenergic agonist, isoproterenol, increased cell proliferation in HPDE6-c7 cells. Western blot and cyclic AMP assays demonstrated that NNK treatments also resulted in: (1) transactivation of the epidermal growth factor receptor, EGFR, (2) an increase in intracellular cyclic AMP accumulation, and (3) phosphorylation of mitogen-activated protein kinase, Erk1/2. The proliferative response to NNK and isoproterenol were inhibited by the use of beta-blockers (propranolol), and the inhibitors of adenylyl cyclase (SQ 22536), EGFR-specific tyrosine kinase (AG 1478) and Erk (PD 98059).
Conclusion: These findings suggest that the NNK -mediated beta-adrenergic receptor transactivation of the EGFR and phosphorylation of Erk1/2 in immortalized human pancreatic duct epithelial cells as a novel mechanism might contribute to the development of tobacco-associated pancreatic carcinogenesis.
Figures
References
-
- Miller BA, Silverman DT, Kaplan R (1993) Pancreas. Cancer Statistics Review: 1973–1990. In: Miller BA, Ries LA, Hanky BF et al (eds) Bethesda, NIH Publication #93–2789, NCI
-
- Gold EB, Cameron JL (1993) Chronic pancreatitis and the risk of pancreatic cancer. N Engl J Med 323:1485–1486 - PubMed
-
- Lowenfels AB, Maisonneuve P, Cavalline G et al (1993) The International Pancreatic Study Group. Pancreatitis and risk of pancreatic cancer. N Engl J Med 328:1433–1437 - PubMed
-
- Hecht SS (1996) Recent studies on the mechanisms of bioactivation and detoxification of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific lung carcinogen. CRC Cri Rev Toxicol 26:163–181 - PubMed
-
- Belinsky A, Devereux TR, Maronpot RR, Stoner GD, Anderson MW (1989) The relationship between the formation of promutagenic adducts and the activation of the K-ras protooncogene in lung tumors from A/J mice treated with nitrosamines. Cancer Res 40:5305–5311 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
