

Gene expression changes during the development of acute lung injury: role of transforming growth factor beta
- PMID: 16100012
- PMCID: PMC2718437
- DOI: 10.1164/rccm.200502-286OC
Gene expression changes during the development of acute lung injury: role of transforming growth factor beta
Abstract
Rationale: Acute lung injury can occur from multiple causes, resulting in high mortality. The pathophysiology of nickel-induced acute lung injury in mice is remarkably complex, and the molecular mechanisms are uncertain.
Objectives: To integrate molecular pathways and investigate the role of transforming growth factor beta (TGF-beta) in acute lung injury in mice.
Methods: cDNA microarray analyses were used to identify lung gene expression changes after nickel exposure. MAPPFinder analysis of the microarray data was used to determine significantly altered molecular pathways. TGF-beta1 protein in bronchoalveolar lavage fluid, as well as the effect of inhibition of TGF-beta, was assessed in nickel-exposed mice. The effect of TGF-beta on surfactant-associated protein B (Sftpb) promoter activity was measured in mouse lung epithelial cells.
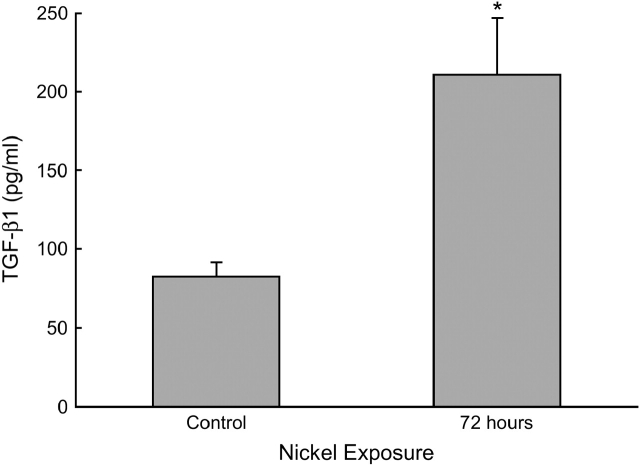
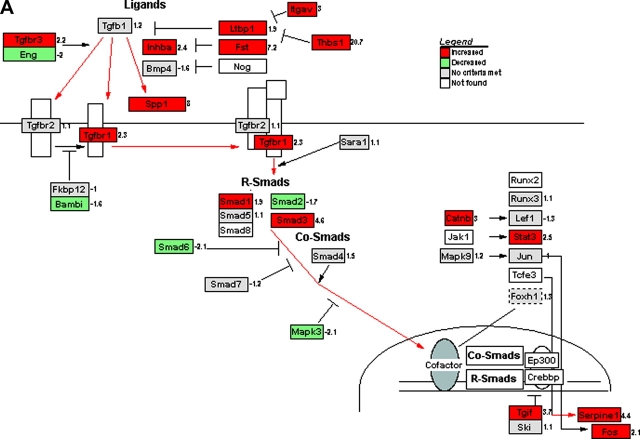
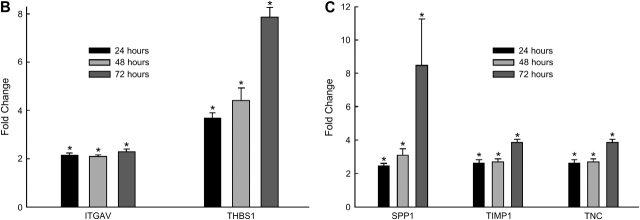
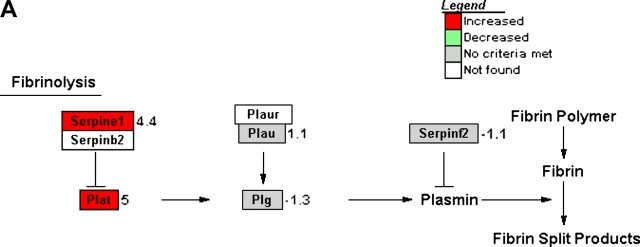
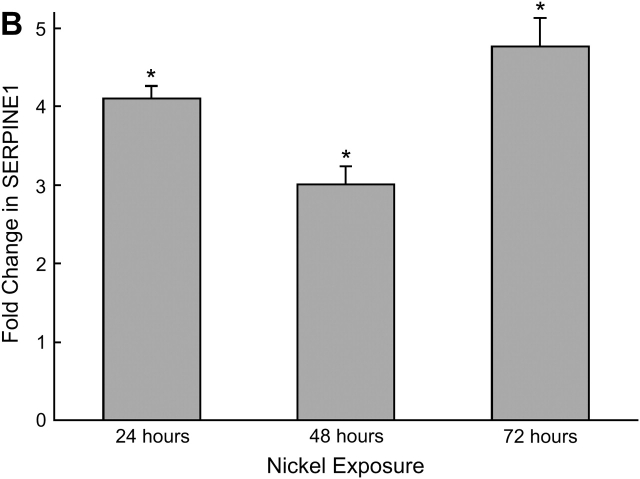
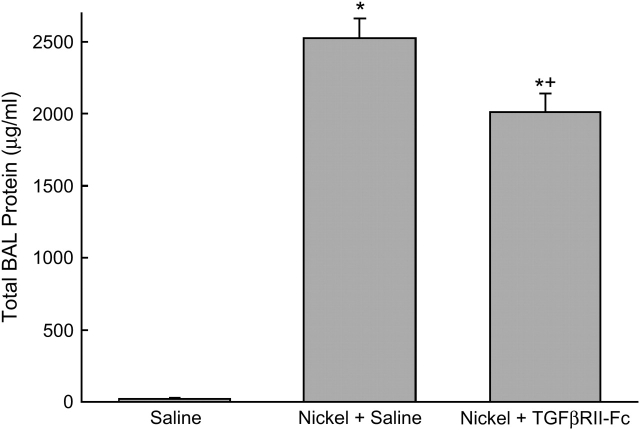
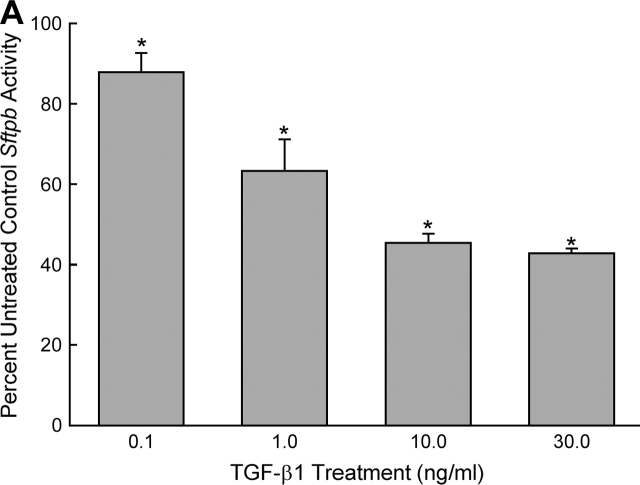
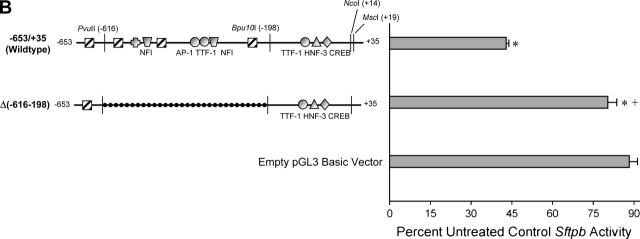
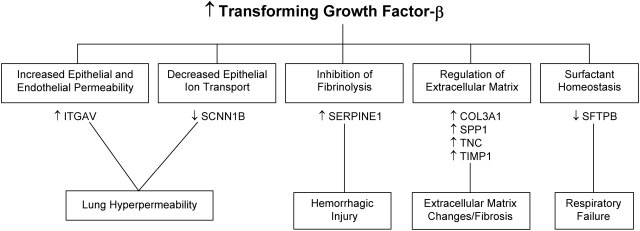
Measurements and main results: Genes that decreased the most after nickel exposure play important roles in lung fluid absorption or surfactant and phospholipid synthesis, and genes that increased the most were involved in TGF-beta signaling. MAPPFinder analysis further established TGF-beta signaling to be significantly altered. TGF-beta-inducible genes involved in the regulation of extracellular matrix function and fibrinolysis were significantly increased after nickel exposure, and TGF-beta1 protein was also increased in the lavage fluid. Pharmacologic inhibition of TGF-beta attenuated nickel-induced protein in bronchoalveolar lavage. In addition, treatment with TGF-beta1 dose-dependently repressed Sftpb promoter activity in vitro, and a novel TGF-beta-responsive region in the Sftpb promoter was identified.
Conclusions: These data suggest that TGF-beta acts as a central mediator of acute lung injury through the alteration of several different molecular pathways.
Figures
Similar articles
-
Keratinocyte growth factor expression is suppressed in early acute lung injury/acute respiratory distress syndrome by smad and c-Abl pathways.Crit Care Med. 2009 May;37(5):1678-84. doi: 10.1097/CCM.0b013e31819fc81a. Crit Care Med. 2009. PMID: 19325470
-
Transforming growth factor-beta: a mediator of cell regulation in acute respiratory distress syndrome.Crit Care Med. 2003 Apr;31(4 Suppl):S258-64. doi: 10.1097/01.CCM.0000057901.92381.75. Crit Care Med. 2003. PMID: 12682450 Review.
-
Surfactant-associated protein B is critical to survival in nickel-induced injury in mice.Am J Respir Cell Mol Biol. 2009 Aug;41(2):226-36. doi: 10.1165/rcmb.2008-0317OC. Epub 2009 Jan 8. Am J Respir Cell Mol Biol. 2009. PMID: 19131640 Free PMC article.
-
Inhibition of nitric oxide restores surfactant gene expression following nickel-induced acute lung injury.Am J Respir Cell Mol Biol. 2003 Feb;28(2):188-98. doi: 10.1165/rcmb.2002-0077OC. Am J Respir Cell Mol Biol. 2003. PMID: 12540486
-
Functional genomics of oxidant-induced lung injury.Adv Exp Med Biol. 2001;500:479-87. doi: 10.1007/978-1-4615-0667-6_73. Adv Exp Med Biol. 2001. PMID: 11764985 Review.
Cited by
-
Oxytocin: A Shield against Radiation-Induced Lung Injury in Rats.Tomography. 2024 Aug 29;10(9):1342-1353. doi: 10.3390/tomography10090101. Tomography. 2024. PMID: 39330747 Free PMC article.
-
The Role of TGF-β in the Association Between Primary Graft Dysfunction and Bronchiolitis Obliterans Syndrome.Am J Transplant. 2016 Feb;16(2):640-9. doi: 10.1111/ajt.13475. Epub 2015 Oct 13. Am J Transplant. 2016. PMID: 26461171 Free PMC article. Clinical Trial.
-
Enolase 1 (ENO1) and protein disulfide-isomerase associated 3 (PDIA3) regulate Wnt/β-catenin-driven trans-differentiation of murine alveolar epithelial cells.Dis Model Mech. 2015 Aug 1;8(8):877-90. doi: 10.1242/dmm.019117. Epub 2015 May 14. Dis Model Mech. 2015. PMID: 26035385 Free PMC article.
-
Early activation of pulmonary TGF-β1/Smad2 signaling in mice with acute pancreatitis-associated acute lung injury.Mediators Inflamm. 2014;2014:148029. doi: 10.1155/2014/148029. Epub 2014 Feb 12. Mediators Inflamm. 2014. PMID: 24688224 Free PMC article.
-
Unlocking the protective potential of the angiotensin type 2 receptor (AT2R) in acute lung injury and age-related pulmonary dysfunction.Biochem Pharmacol. 2024 Feb;220:115978. doi: 10.1016/j.bcp.2023.115978. Epub 2023 Dec 9. Biochem Pharmacol. 2024. PMID: 38081369 Free PMC article.
References
-
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000;342:1334–1349. - PubMed
-
- Lewis JF, Jobe AH. Surfactant and the adult respiratory distress syndrome. Am Rev Respir Dis 1993;147:218–233. - PubMed
-
- Grande JP. Role of transforming growth factor-beta in tissue injury and repair. Proc Soc Exp Biol Med 1997;214:27–40. - PubMed
-
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med 1999;341:738–746. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
