

Using multiple alignments to improve seeded local alignment algorithms
- PMID: 16100379
- PMCID: PMC1185574
- DOI: 10.1093/nar/gki767
Using multiple alignments to improve seeded local alignment algorithms
Abstract
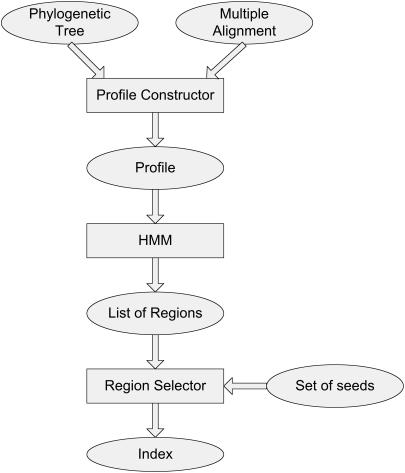
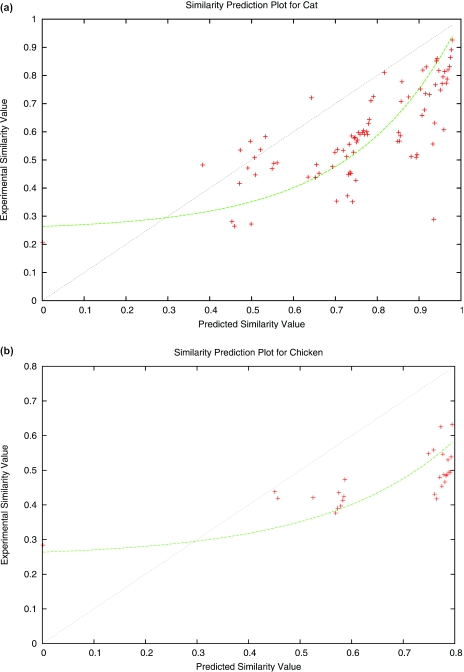
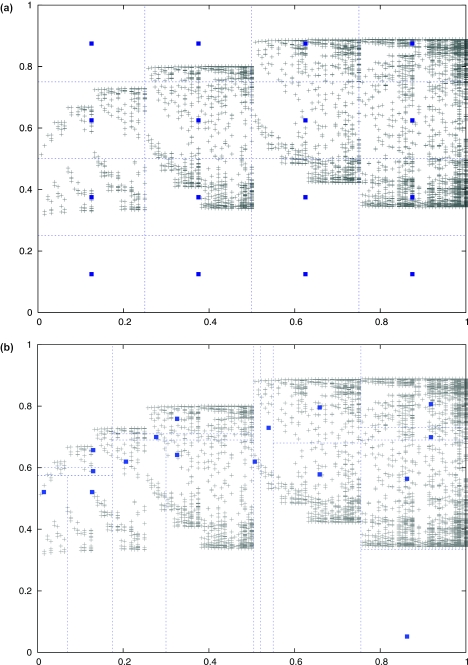
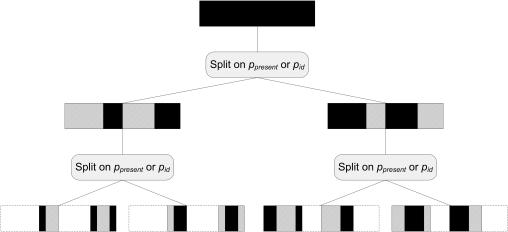
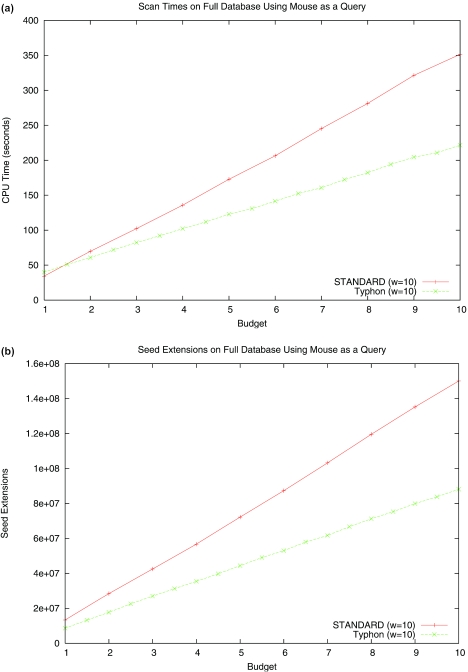
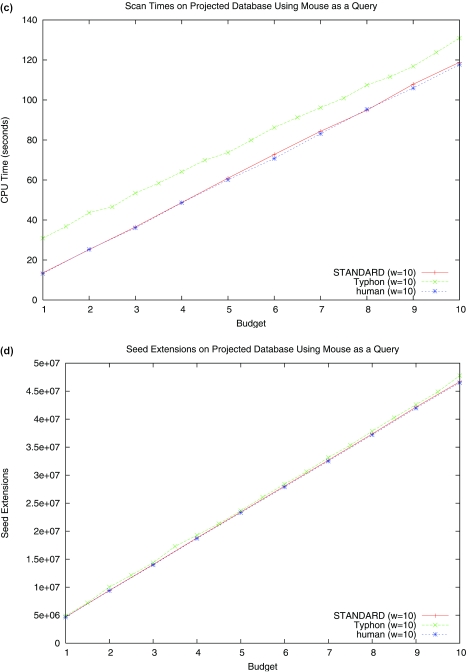
Multiple alignments among genomes are becoming increasingly prevalent. This trend motivates the development of tools for efficient homology search between a query sequence and a database of multiple alignments. In this paper, we present an algorithm that uses the information implicit in a multiple alignment to dynamically build an index that is weighted most heavily towards the promising regions of the multiple alignment. We have implemented Typhon, a local alignment tool that incorporates our indexing algorithm, which our test results show to be more sensitive than algorithms that index only a sequence. This suggests that when applied on a whole-genome scale, Typhon should provide improved homology searches in time comparable to existing algorithms.
Figures

Similar articles
-
Multiple whole-genome alignments without a reference organism.Genome Res. 2009 Apr;19(4):682-9. doi: 10.1101/gr.081778.108. Epub 2009 Jan 28. Genome Res. 2009. PMID: 19176791 Free PMC article.
-
Uncertainty in homology inferences: assessing and improving genomic sequence alignment.Genome Res. 2008 Feb;18(2):298-309. doi: 10.1101/gr.6725608. Epub 2007 Dec 11. Genome Res. 2008. PMID: 18073381 Free PMC article.
-
GSAlign: an efficient sequence alignment tool for intra-species genomes.BMC Genomics. 2020 Feb 24;21(1):182. doi: 10.1186/s12864-020-6569-1. BMC Genomics. 2020. PMID: 32093618 Free PMC article.
-
Evolution at the nucleotide level: the problem of multiple whole-genome alignment.Hum Mol Genet. 2006 Apr 15;15 Spec No 1:R51-6. doi: 10.1093/hmg/ddl056. Hum Mol Genet. 2006. PMID: 16651369 Review.
-
Sequence alignment in molecular biology.J Comput Biol. 1998 Summer;5(2):173-96. doi: 10.1089/cmb.1998.5.173. J Comput Biol. 1998. PMID: 9672827 Review.
Cited by
-
The distance-profile representation and its application to detection of distantly related protein families.BMC Bioinformatics. 2005 Nov 29;6:282. doi: 10.1186/1471-2105-6-282. BMC Bioinformatics. 2005. PMID: 16316461 Free PMC article.
-
MapToGenome: a comparative genomic tool that aligns transcript maps to sequenced genomes.Evol Bioinform Online. 2007 Feb 14;3:15-25. Evol Bioinform Online. 2007. PMID: 19430601 Free PMC article.
References
-
- Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
