

Hepatitis A virus suppresses RIG-I-mediated IRF-3 activation to block induction of beta interferon
- PMID: 16103148
- PMCID: PMC1193608
- DOI: 10.1128/JVI.79.17.10968-10977.2005
Hepatitis A virus suppresses RIG-I-mediated IRF-3 activation to block induction of beta interferon
Abstract
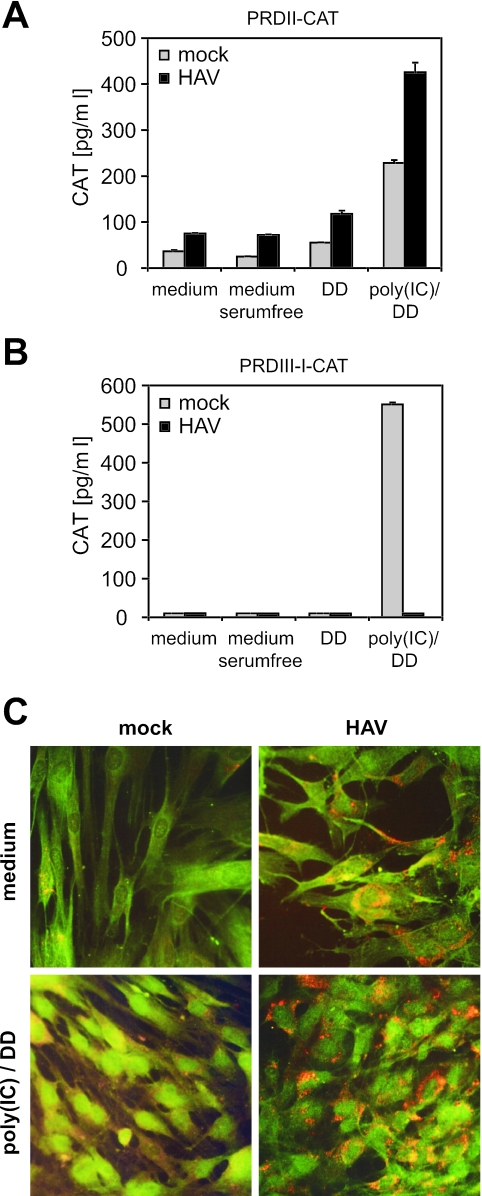
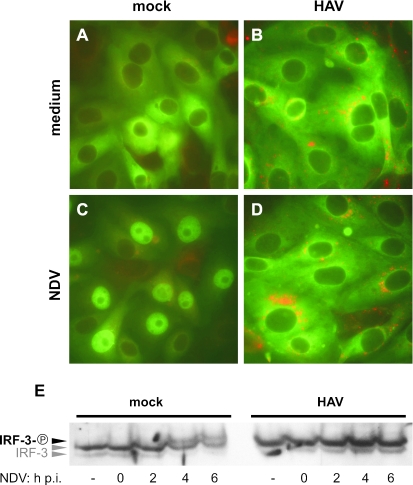
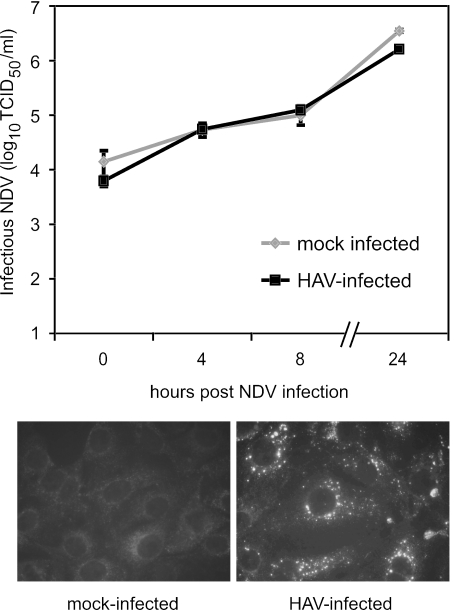
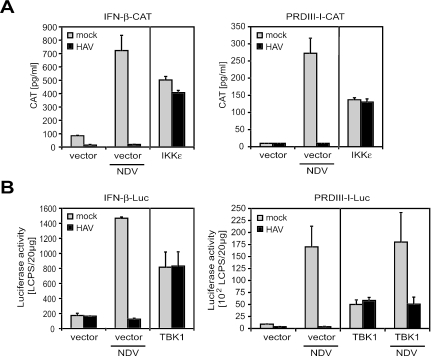
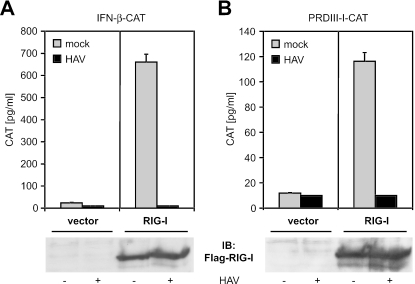
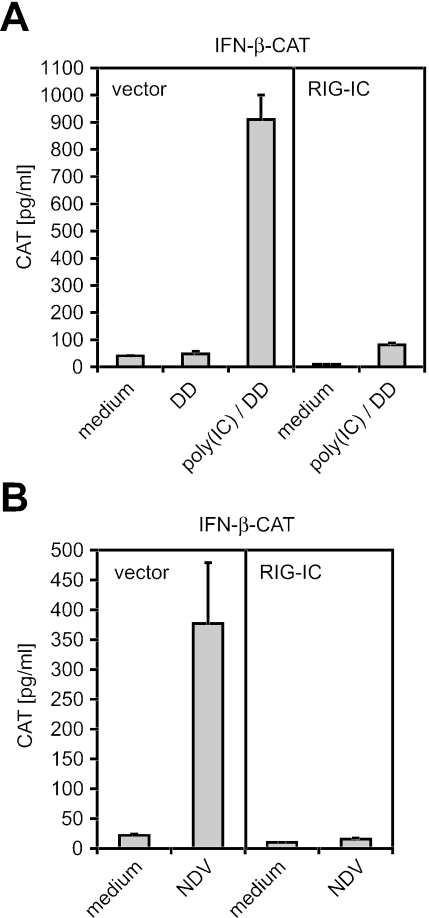
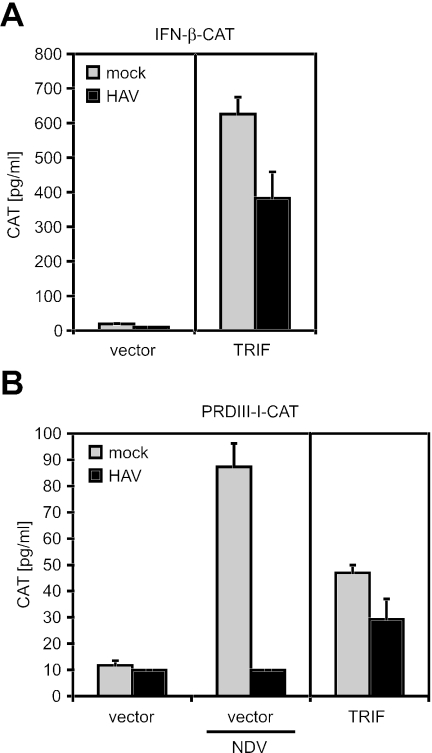
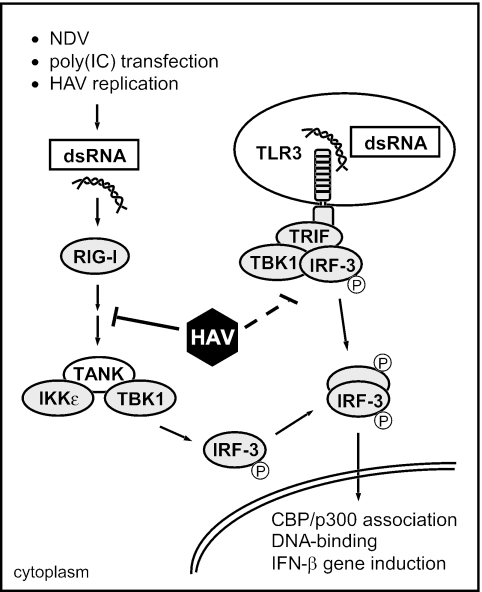
Hepatitis A virus (HAV) antagonizes the innate immune response by inhibition of double-stranded RNA (dsRNA)-induced beta interferon (IFN-beta) gene expression. In this report, we show that this is due to an interaction of HAV with the intracellular dsRNA-induced retinoic acid-inducible gene I (RIG-I)-mediated signaling pathway upstream of the kinases responsible for interferon regulatory factor 3 (IRF-3) phosphorylation (TBK1 and IKKepsilon). In consequence, IRF-3 is not activated for nuclear translocation and gene induction. In addition, we found that HAV reduces TRIF (TIR domain-containing adaptor inducing IFN-beta)-mediated IRF-3 activation, which is part of the Toll-like receptor 3 signaling pathway. As IRF-3 is necessary for IFN-beta transcription, inhibition of this factor results in efficient suppression of IFN-beta synthesis. This ability of HAV seems to be of considerable importance for HAV replication, as HAV is not resistant to IFN-beta, and it may allow the virus to establish infection and preserve the sites of virus production in later stages of the infection.
Figures
Similar articles
-
Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon.J Virol. 2005 Apr;79(7):3969-78. doi: 10.1128/JVI.79.7.3969-3978.2005. J Virol. 2005. PMID: 15767399 Free PMC article.
-
Induction of IRF-3 and IRF-7 phosphorylation following activation of the RIG-I pathway.Cell Mol Biol (Noisy-le-grand). 2006 May 15;52(1):17-28. Cell Mol Biol (Noisy-le-grand). 2006. PMID: 16914100
-
Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling.J Immunol. 2003 Oct 15;171(8):4304-10. doi: 10.4049/jimmunol.171.8.4304. J Immunol. 2003. PMID: 14530355
-
Hepatitis A and hepatitis C viruses: divergent infection outcomes marked by similarities in induction and evasion of interferon responses.Semin Liver Dis. 2010 Nov;30(4):319-32. doi: 10.1055/s-0030-1267534. Epub 2010 Oct 19. Semin Liver Dis. 2010. PMID: 20960373 Review.
-
The host type I interferon response to viral and bacterial infections.Cell Res. 2005 Jun;15(6):407-22. doi: 10.1038/sj.cr.7290309. Cell Res. 2005. PMID: 15987599 Review.
Cited by
-
Theiler's virus strain-dependent induction of innate immune responses in RAW264.7 macrophages and its influence on viral clearance versus viral persistence.J Neurovirol. 2007;13(1):47-55. doi: 10.1080/13550280601145357. J Neurovirol. 2007. PMID: 17454448
-
Host Innate Immunity Against Hepatitis Viruses and Viral Immune Evasion.Front Microbiol. 2021 Nov 3;12:740464. doi: 10.3389/fmicb.2021.740464. eCollection 2021. Front Microbiol. 2021. PMID: 34803956 Free PMC article. Review.
-
Hepatocyte GPCR signaling regulates IRF3 to control hepatic stellate cell transdifferentiation.Cell Commun Signal. 2024 Jan 17;22(1):48. doi: 10.1186/s12964-023-01416-6. Cell Commun Signal. 2024. PMID: 38233853 Free PMC article.
-
Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway.Mol Immunol. 2008 May;45(10):2839-46. doi: 10.1016/j.molimm.2008.01.028. Epub 2008 Mar 11. Mol Immunol. 2008. PMID: 18336912 Free PMC article.
-
Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor.Proc Natl Acad Sci U S A. 2007 Apr 24;104(17):7253-8. doi: 10.1073/pnas.0611506104. Epub 2007 Apr 16. Proc Natl Acad Sci U S A. 2007. PMID: 17438296 Free PMC article.
References
-
- Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499-511. - PubMed
-
- Alexopoulou, L., A. C. Holt, R. Medzhitov, and R. A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature 413:732-738. - PubMed
-
- Balachandran, S., E. Thomas, and G. N. Barber. 2004. A FADD-dependent innate immune mechanism in mammalian cells. Nature 432:401-405. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
