

Human immunodeficiency virus type 1 Vpr-dependent cell cycle arrest through a mitogen-activated protein kinase signal transduction pathway
- PMID: 16103188
- PMCID: PMC1193619
- DOI: 10.1128/JVI.79.17.11366-11381.2005
Human immunodeficiency virus type 1 Vpr-dependent cell cycle arrest through a mitogen-activated protein kinase signal transduction pathway
Abstract
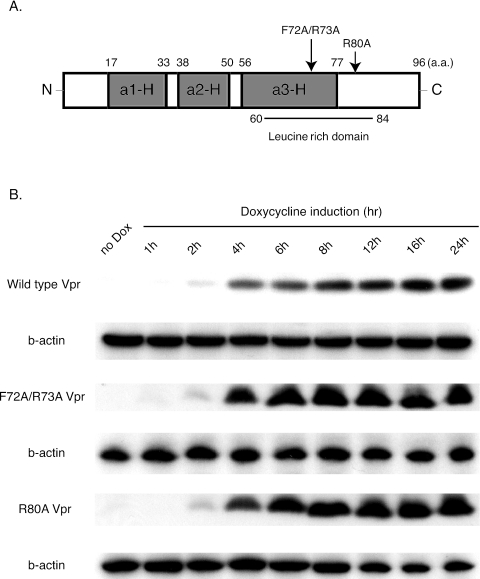
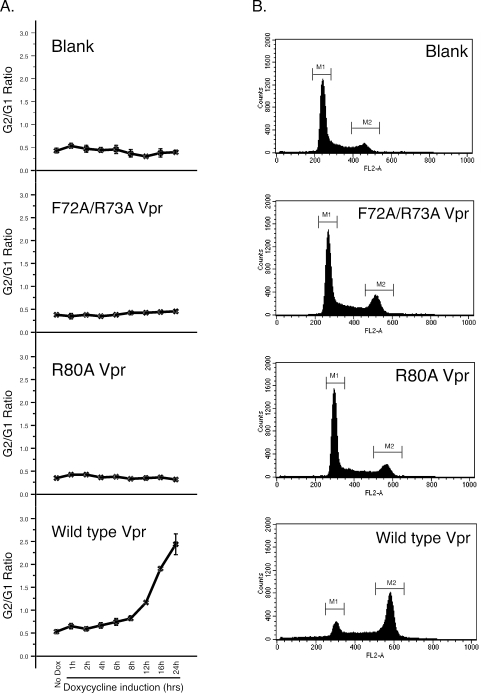
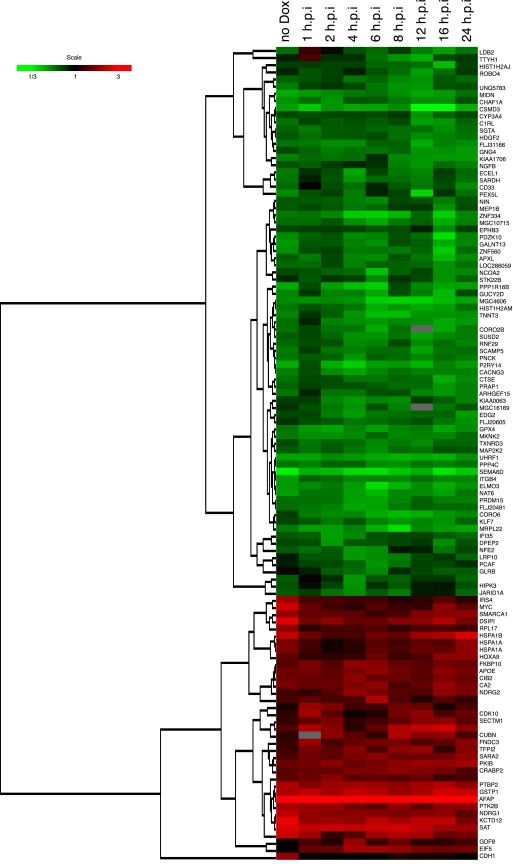
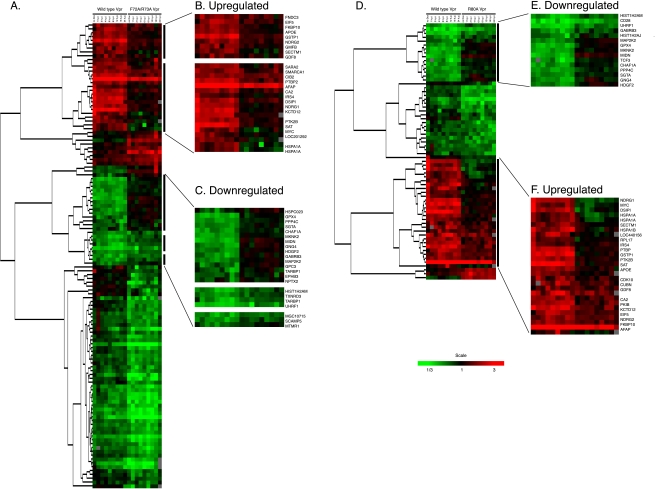
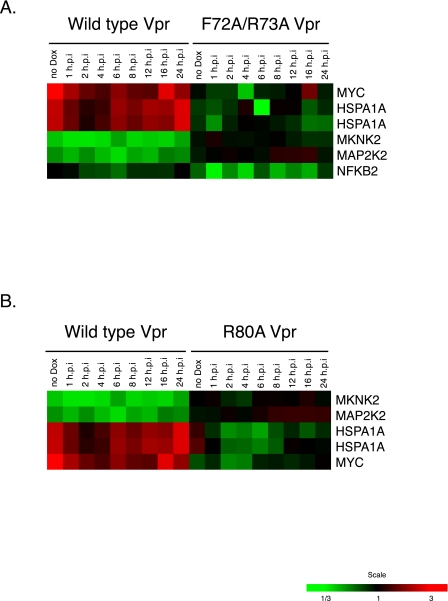
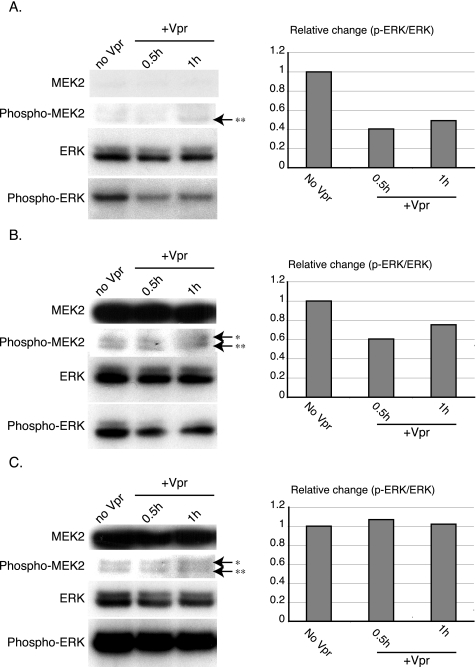
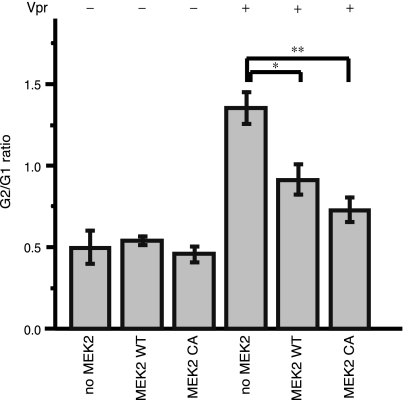
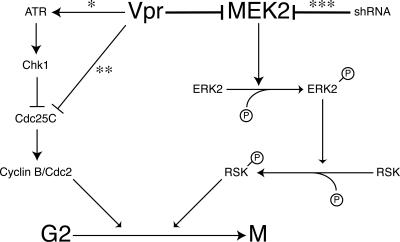
The human immunodeficiency virus type 1 (HIV-1) Vpr protein has important functions in advancing HIV pathogenesis via several effects on the host cell. Vpr mediates nuclear import of the preintegration complex, induces host cell apoptosis, and inhibits cell cycle progression at G(2), which increases HIV gene expression. Some of Vpr's activities have been well described, but some functions, such as cell cycle arrest, are not yet completely characterized, although components of the ATR DNA damage repair pathway and the Cdc25C and Cdc2 cell cycle control mechanisms clearly play important roles. We investigated the mechanisms underlying Vpr-mediated cell cycle arrest by examining global cellular gene expression profiles in cell lines that inducibly express wild-type and mutant Vpr proteins. We found that Vpr expression is associated with the down-regulation of genes in the MEK2-ERK pathway and with decreased phosphorylation of the MEK2 effector protein ERK. Exogenous provision of excess MEK2 reverses the cell cycle arrest associated with Vpr, confirming the involvement of the MEK2-ERK pathway in Vpr-mediated cell cycle arrest. Vpr therefore appears to arrest the cell cycle at G(2)/M through two different mechanisms, the ATR mechanism and a newly described MEK2 mechanism. This redundancy suggests that Vpr-mediated cell cycle arrest is important for HIV replication and pathogenesis. Our findings additionally reinforce the idea that HIV can optimize the host cell environment for viral replication.
Figures
References
-
- Abbott, D. W., and J. T. Holt. 1999. Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J. Biol. Chem. 274:2732-2742. - PubMed
-
- Chowdhury, I. H., X. F. Wang, N. R. Landau, M. L. Robb, V. R. Polonis, D. L. Birx, and J. H. Kim. 2003. HIV-1 Vpr activates cell cycle inhibitor p21/Waf1/Cip1: a potential mechanism of G2/M cell cycle arrest. Virology 305:371-377. - PubMed
-
- Cohen, E. A., E. F. Terwilliger, Y. Jalinoos, J. Proulx, J. G. Sodroski, and W. A. Haseltine. 1990. Identification of HIV-1 vpr product and function. J. Acquir. Immune Defic. Syndr. 3:11-18. - PubMed
-
- Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935-944. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
