

Ab initio prediction of transcription factor targets using structural knowledge
- PMID: 16103898
- PMCID: PMC1183507
- DOI: 10.1371/journal.pcbi.0010001
Ab initio prediction of transcription factor targets using structural knowledge
Abstract
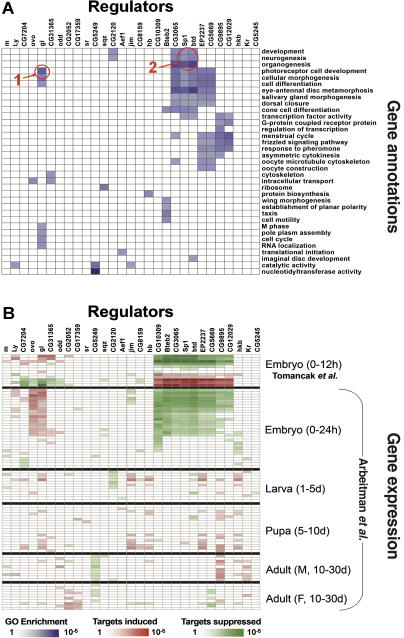
Current approaches for identification and detection of transcription factor binding sites rely on an extensive set of known target genes. Here we describe a novel structure-based approach applicable to transcription factors with no prior binding data. Our approach combines sequence data and structural information to infer context-specific amino acid-nucleotide recognition preferences. These are used to predict binding sites for novel transcription factors from the same structural family. We demonstrate our approach on the Cys(2)His(2) Zinc Finger protein family, and show that the learned DNA-recognition preferences are compatible with experimental results. We use these preferences to perform a genome-wide scan for direct targets of Drosophila melanogaster Cys(2)His(2) transcription factors. By analyzing the predicted targets along with gene annotation and expression data we infer the function and activity of these proteins.
Conflict of interest statement
Figures





References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
